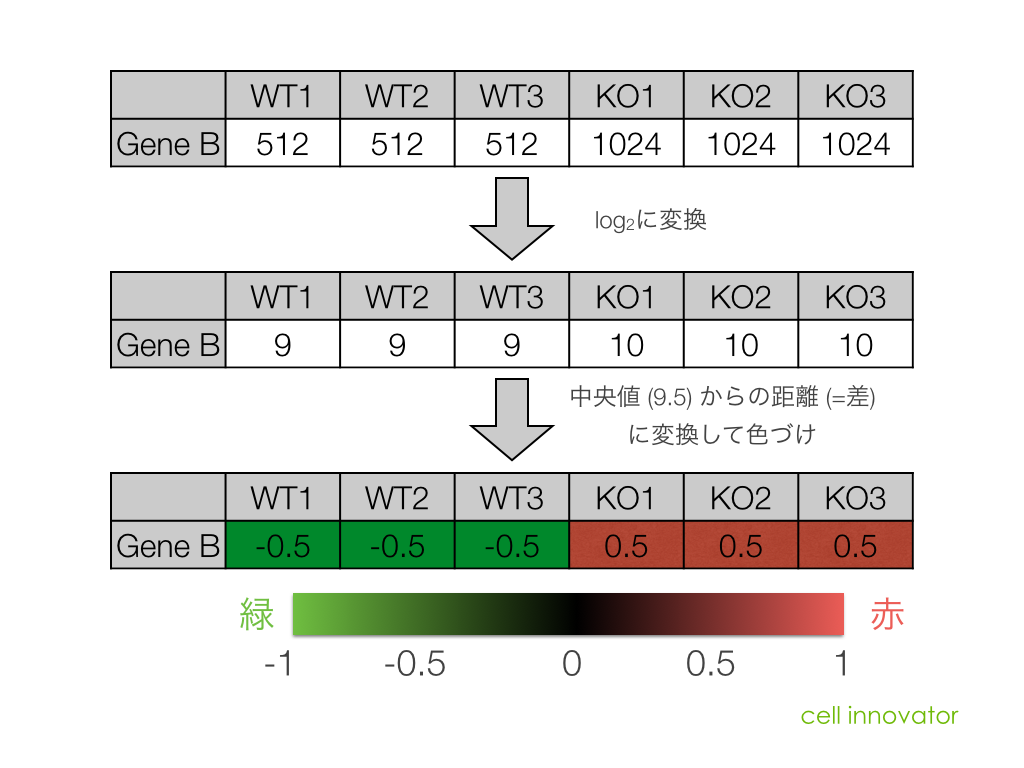
マイクロアレイにおいて、シグナル値の低いものは、信頼性(データの再現性)がよくない場合があります。これは、MAプロットや散布図の形状からも推測されます。(同一条件で比較したサンプルにおいても、シグナル値の低い部分に変動が見られがち。)
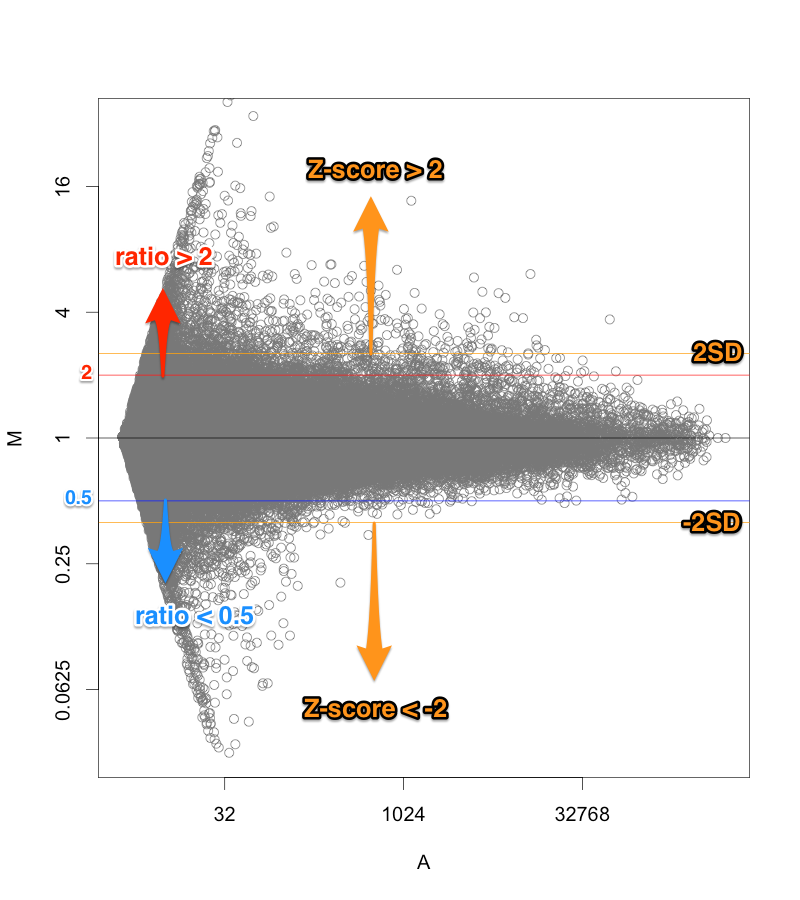
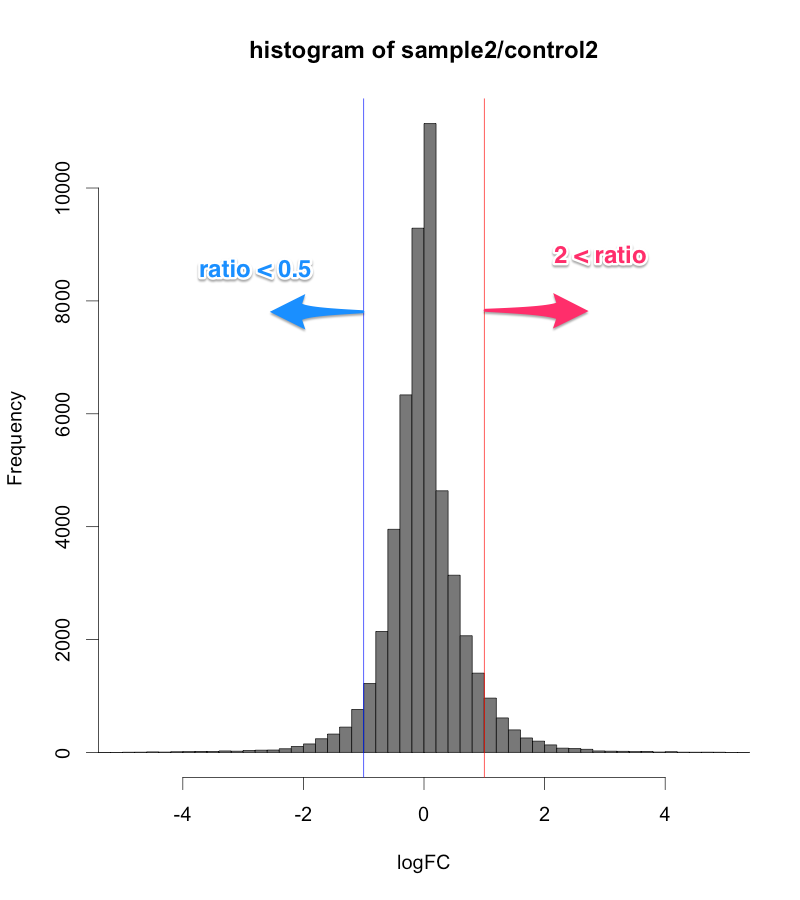
例えば、シグナル値が、1000から10000になったら、ratio=10ですが、10から100になっても同じ ratio=10 です。ratio だけで、発現変動遺伝子を判定すると、特にシグナル値の低い部分に、ratio が高い遺伝子が見つかりがちですが、再現性を考えた場合、少し不安です。
一方、 Z-score で判定した場合も、MA プロットから分かるように、シグナル値の高い部分は、Z-score が高くなりにくい傾向があります。
この2点の問題を解消するために、 Intensity-based Z-score (Intensity-dependent Z-score)* というものがあります。(以前から提案されているもので、珍しい手法ではありません。)
Intensity-based Z-score
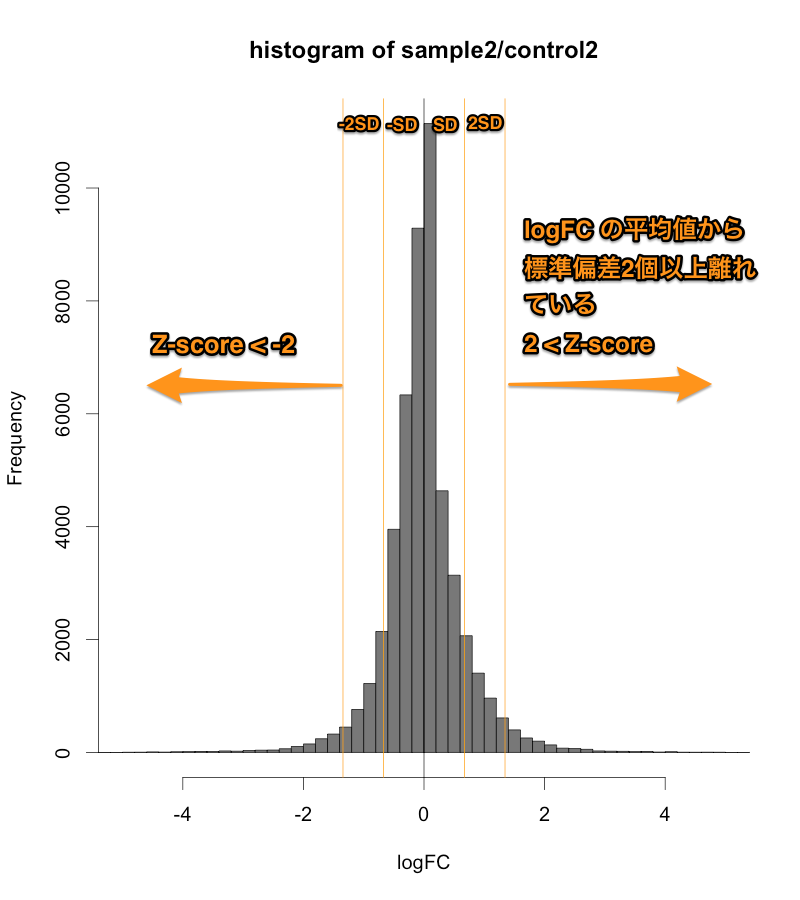
Intensity-based Z-score では、文字通り、Intensity (=シグナル値の強さ) を考慮した Z-score です。マイクロアレイデータをシグナル値の大きさごとに一定の区間に分割して、その区間ごとに logFC の平均値と標準偏差を求めます。そして、それぞれの区間ごとに、Z-score を算出します。
そうすることで、シグナル値の低い部分は、大きい標準偏差 (SD) を用い、一方、シグナル値の高い部分は、小さい SD を用いて Z-score を判定することになります。したがって、シグナル値の低い部分は、通常の Z-score より判定条件が厳しくなり、シグナル値の高い部分は、判定条件が緩くなります。
MAプロットで確認すると、違いがよくわかります。ここでは、便宜的に、シグナル値の大きさによって8区間に分割して、Z-score を算出しています(図下)。(8つという区間に必然性はありません。もう少し細く区切ってもよいです。)また、Intensity-based Z-score を使用した図は、事前に使用した2サンプルのフラグが、共にA(absent: 未検出)のプローブを除外しています。
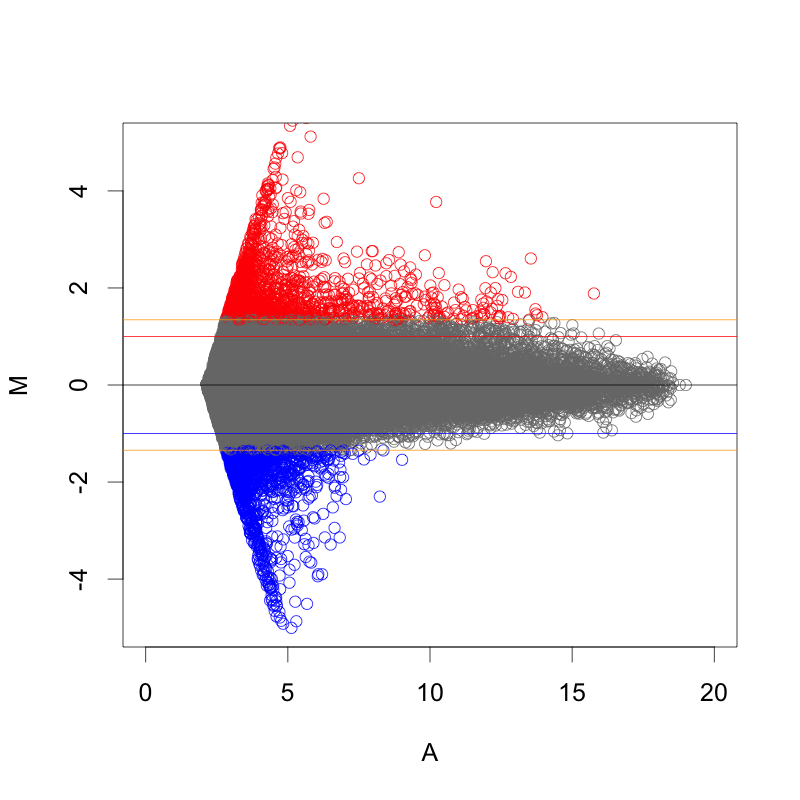

図形的には、変動している遺伝子を外側から数パーセント取るという形になります。単純に ratio で判定するよりも慎重な方法といえるでしょう。しかしながら、これも万能な手法というわけではありません。可能であれば、繰り返しサンプル (replicates) が、3サンプル以上 (n=3以上) あって、検定などの手法が使える方が望ましいことには変わりません。
参考
* Quackenbush. Microarray data normalization and transformation. Nat Genet (2002) vol. 32 Suppl pp. 496-501.