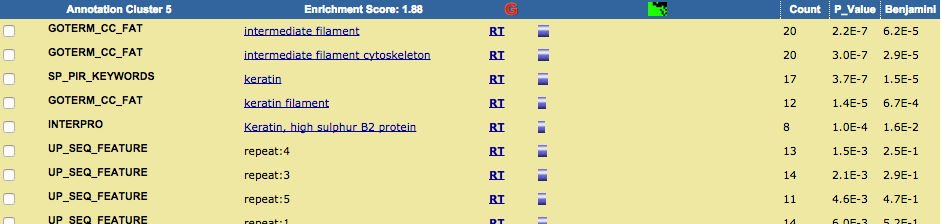
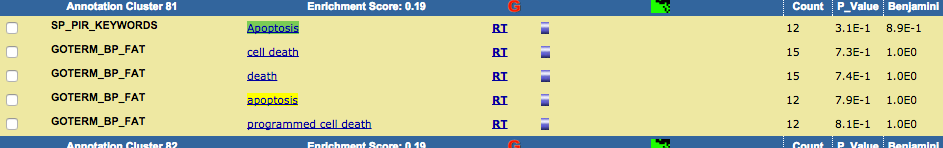
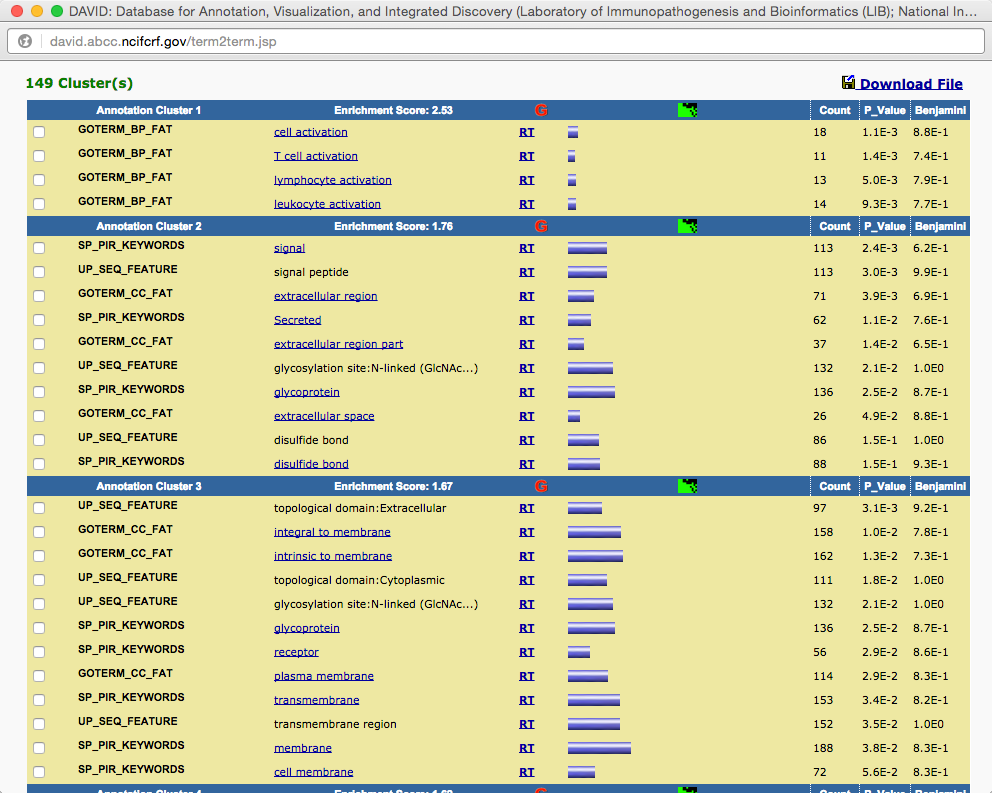
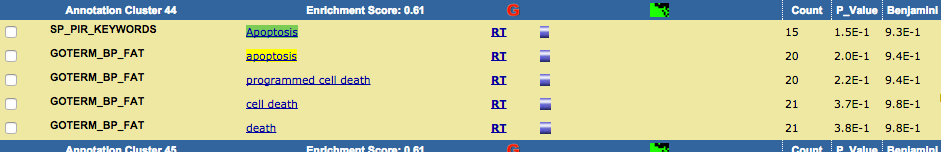
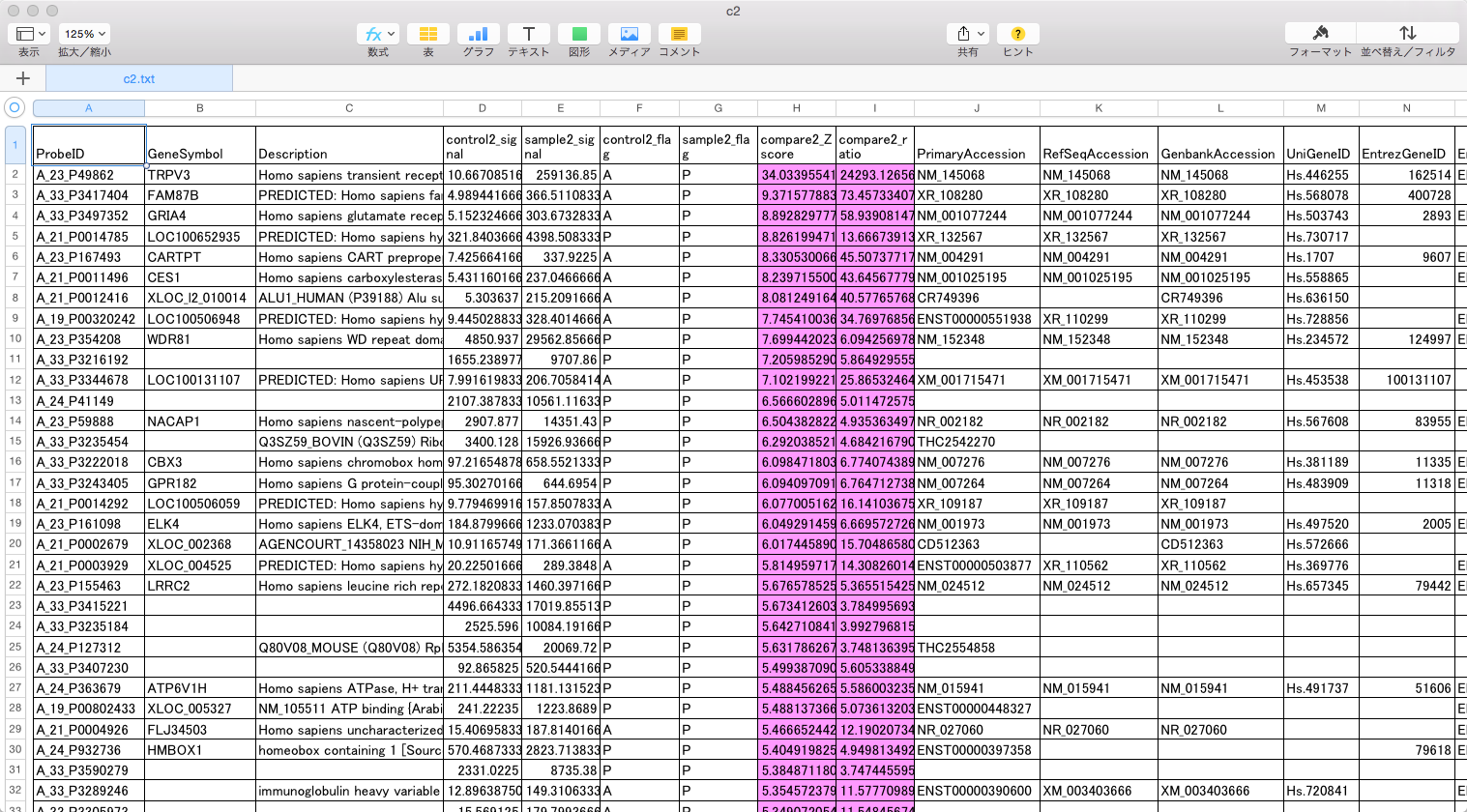
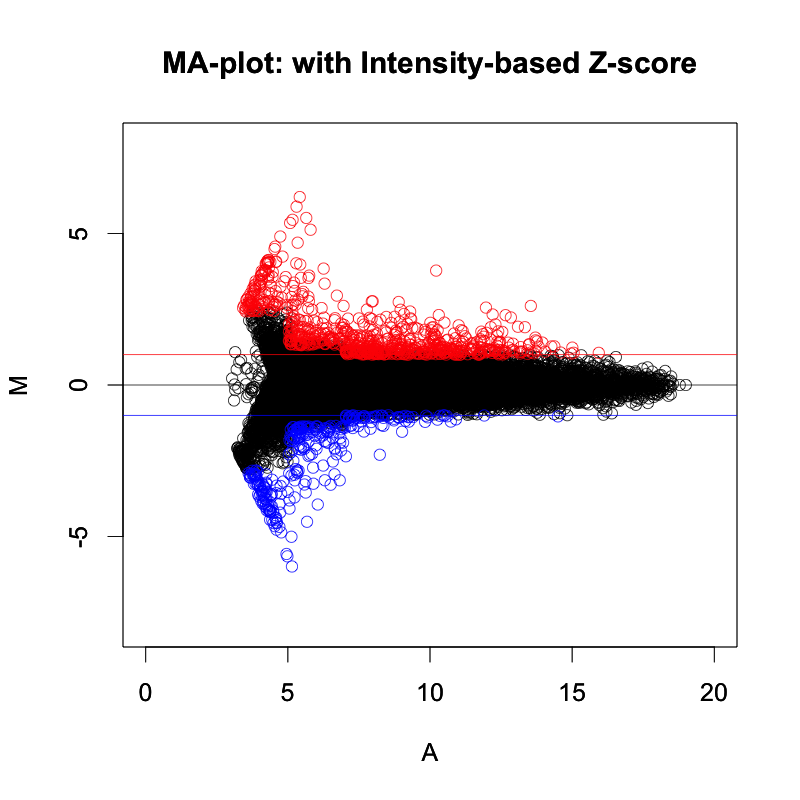
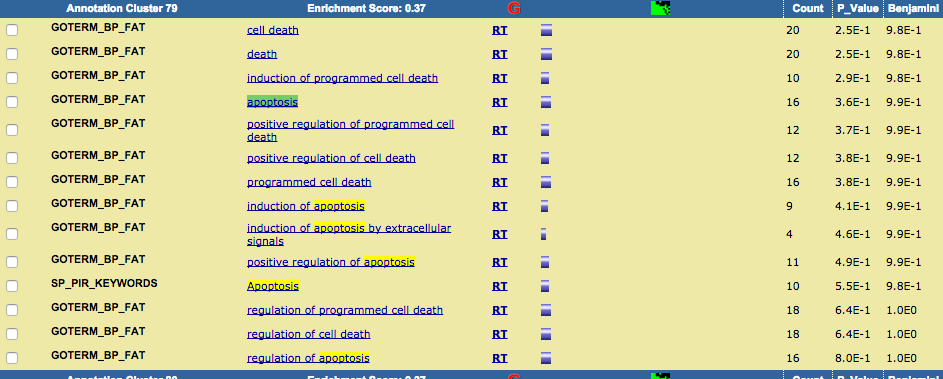
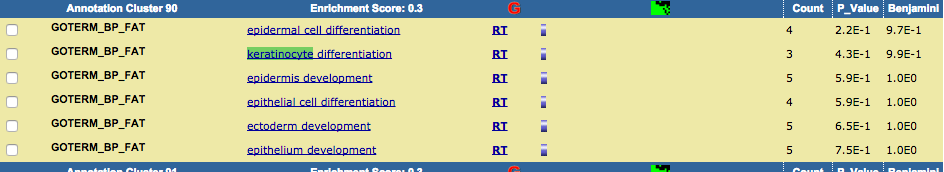
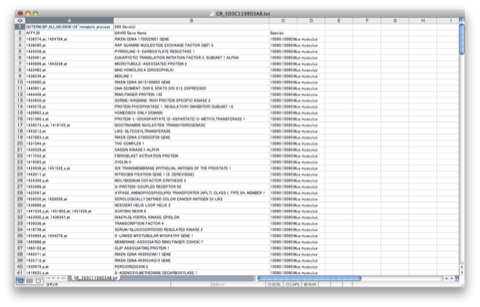
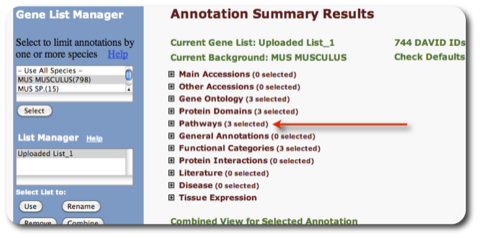
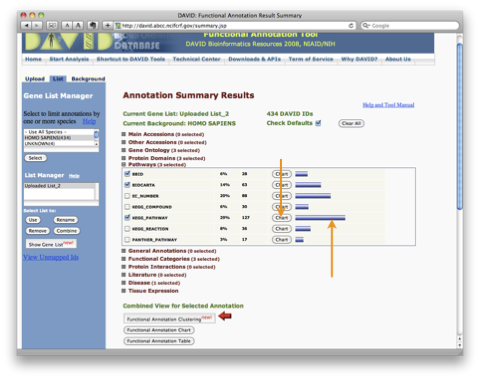
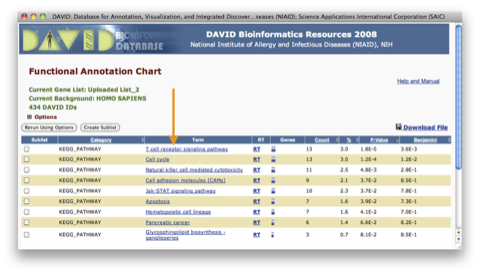
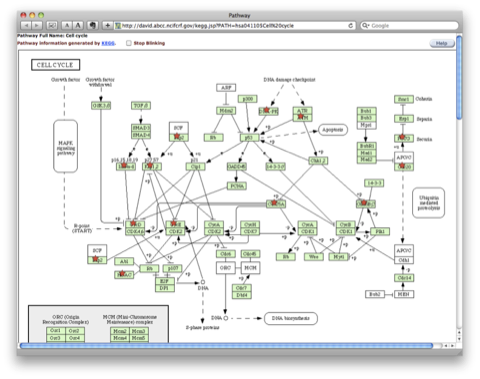
TRPV3 dysfunction may increase apoptotic activity, inhibit keratinocyte differentiation and disturb the intricate balance between proliferation and differentiation state of keratinocytes in the skin.
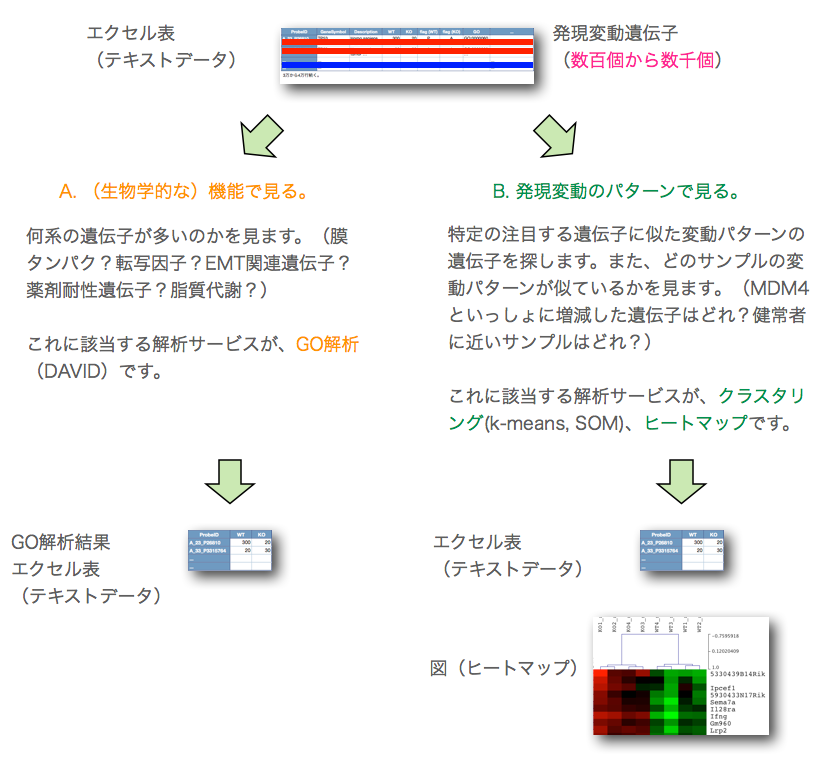
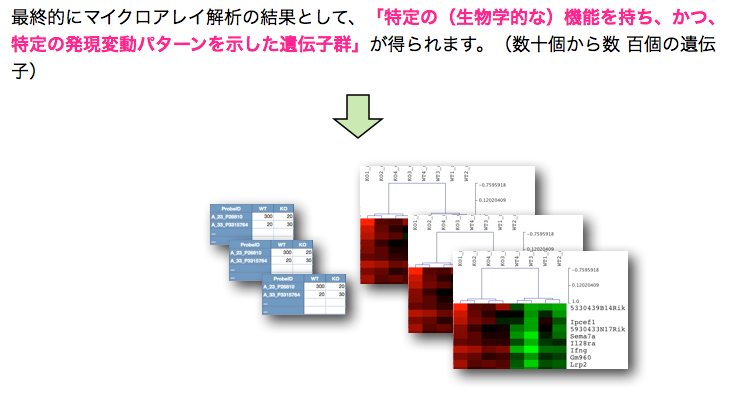
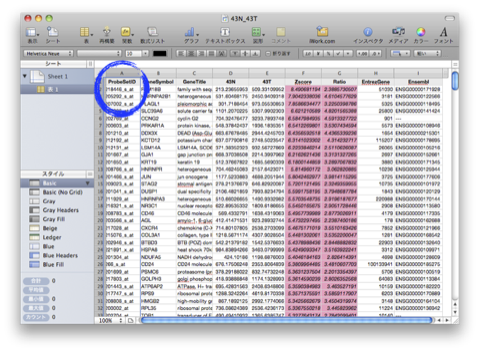
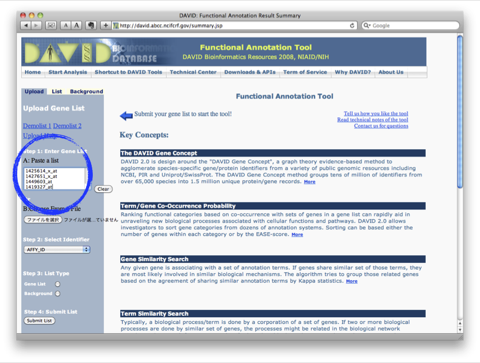
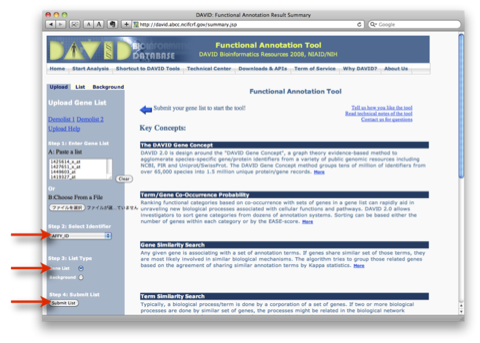
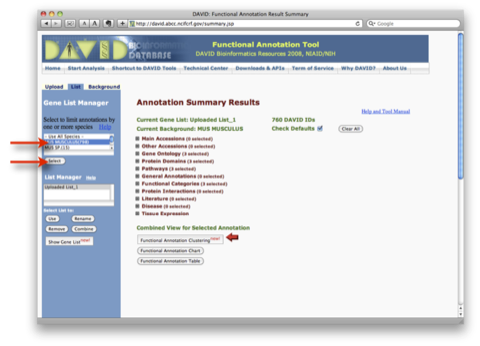
このA., B. の2通りの解析は、独立したものではなく、また、それだけでは終わりません。GO解析の結果をさらにヒートマップで表示して、発現変動変動のパターンを確認するケース(A. –> B.)や、特定の発現変動パターンの遺伝子を選択して、その機能をGO解析で確認するケース(B. –> A.) が通常です。それぞれの解析を単独で行っても効果的ではありません。(セルイノベーターの解析サービスでは、初めからこれらの解析サービスを含めて提供しています。)
Reference
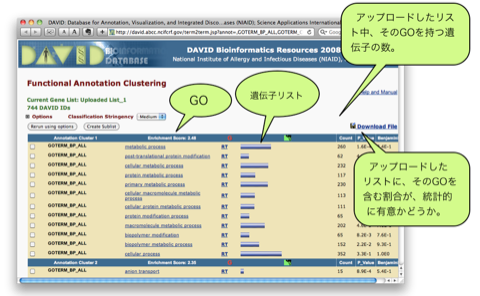
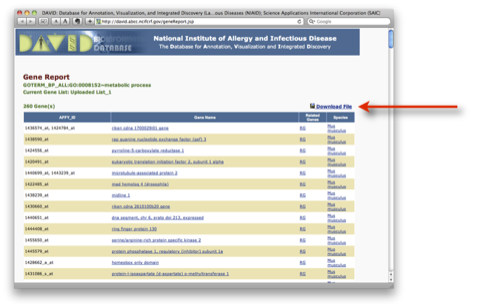
Huang et al. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols (2009) vol. 4 (1) pp. 44-57.