現在の MeV は、オンライン版(ブラウザ上で操作)になっています。
引き続き、オフライン版(=stand alone client)も SourceForge よりダウンロードが可能です。
> https://sourceforge.net/projects/mev-tm4/

現在の MeV は、オンライン版(ブラウザ上で操作)になっています。
引き続き、オフライン版(=stand alone client)も SourceForge よりダウンロードが可能です。
> https://sourceforge.net/projects/mev-tm4/
シグナル値ではなく、ratio でヒートマップを書いて、変動パターンにをチェックすることもあります。
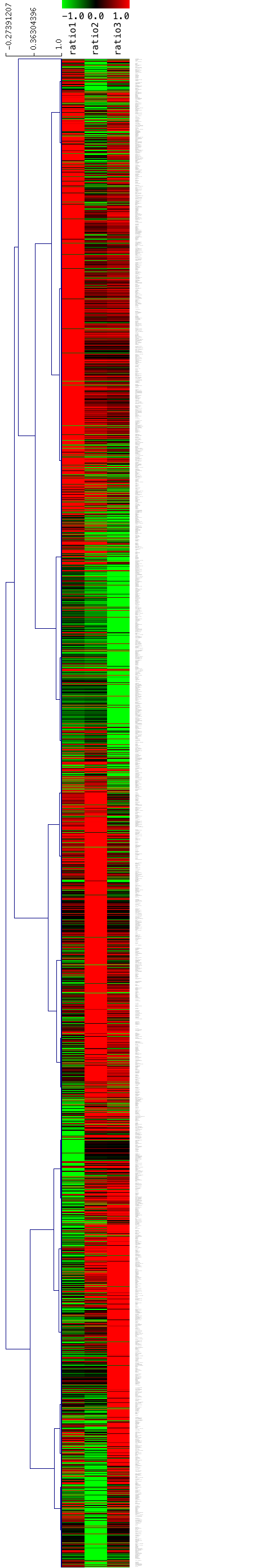
シグナル値ではなく、ratio を用いたヒートマップの例を示します。データとしては、解析例1の3つの比較において、いずれかの比較で変動のあった遺伝子(約2678個)だけを用いています。
左から、3つの比較(ratio1, ratio2, ratio3) を並べています。ratio1 = sample1/control1, ratio2=sample2/control2, ratio3=sample3/control3 となります。
色付けは、ratio < 0.5 を明るい緑、ratio > 2 が明るい赤になるようにしています。
また、図は全体が見えやすいようにリサイズしています。

シグナル値を用いた場合のヒートマップと異なり、ヒートマップにコントロールは表示されません。3列とも赤であれば、3つの比較において、共通に増加した遺伝子だと分かります。逆に3列とも緑であれば、共通に減少した遺伝子です。
変動パターンをチェックしやすい反面、計算していない比較の組み合わせ(例えば、sample2 と sample3 ではどちらが高いか)は読み取るのが難しい場合があります。
クラスタリングを解釈する際に、よく誤解しやすい部分があります。クラスタリングの結果というものは、それほど絶対的なものではありません。
クラスタリングの結果は、どのアルゴリズム(hierarchical, k-means, SOM など)を用いてクラスタリングするかで多少異なりますし、同じアルゴリズムを用いたとしても、パラメーターの設定によっても若干変化します。
また、クラスタリングするときに、全遺伝子を用いるか、変動している遺伝子だけを用いるかでも変わります。
サンプル方向にクラスタリングを行い、似ている順位のツリーを書くためには、各サンプル間の距離を求めることになります。このサンプル間の距離を求める方法は、いくつかあります。例えば、単純に引き算して差を取るだけの場合や、2乗してから差を取る場合、相関係数を用いる場合などです。
MeV では、 Distance Metric Selection の部分で、この距離を求める手法を選択することになります。
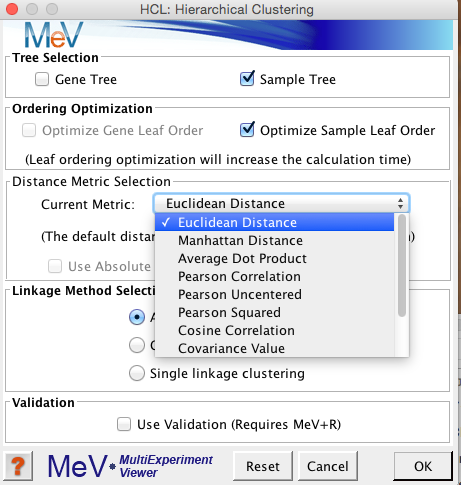
このサンプル間の距離の求め方によって、最終的な結果のツリーの分類は多少異なります。解析例1の全遺伝子(5万プローブ)をサンプル方向のみでクラスタリングした結果を示します。
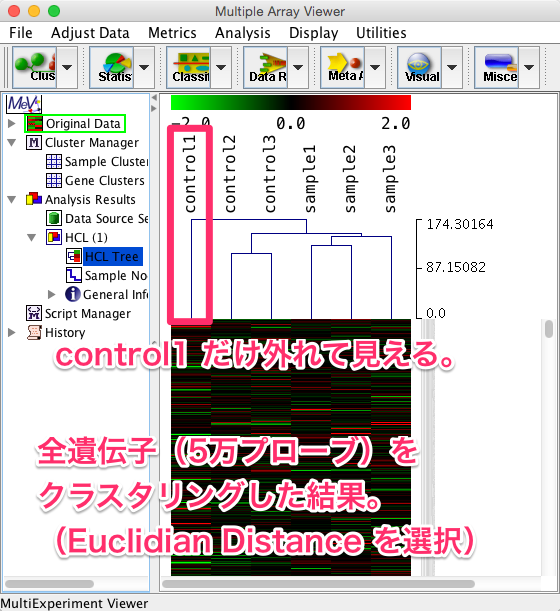
距離を求める手法に Euclidian Distance を選択した場合、control1 だけ外れて(似ていないように)見えます。
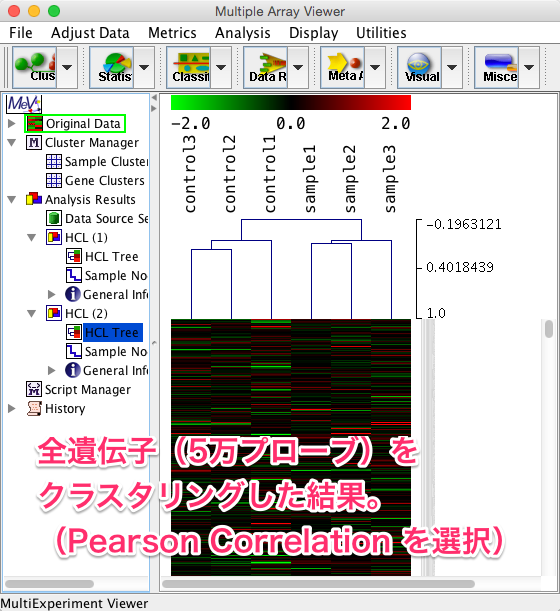
相関係数(Pearson Correlation)を用いて、クラスタリングした場合、control1だけ外れているようには見えません。一般的には、この相関係数を用いることが多いと思います。
上述の全遺伝子を用いてクラスタリングした結果に対して、変動している遺伝子だけをクラスタリングした結果を示します。 全遺伝子を対象とした場合と異なり、 sample2 と sample3 が近いように見えています。

このようにクラスタリングの結果は、必ずしも、絶対的なものではありません。よく論文で図示されていますが、あくまで1例であり、あるパターンに分かれる可能性を示したものにすぎません。(仮説の証明手段ではありません。)
特に全遺伝子でクラスタリングした場合、「sample1 と一番近いのは、 sample2 と sample3 のどちらか?」というような、結果の細部にあまりこだわらないほうがよいでしょう。前述のような計算方法の違いで多少前後する可能性があります。ある意味、当然のことなので、これらの結果に一喜一憂する必要はありません。
(その時、用いたデータセットとパラメーターを使えば、とりあえず、あるパターンに分かれて見えるという程度に理解した方が良いです。結果の解釈には生物学的な裏付けが求められます。)
似ているサンプルを探したい場合は、クラスタリングで見つけることができます。 MeV を用いた解析例を示します。
似ているサンプルを探していたい場合、クラスタリングのアルゴリズムとしては、「階層的クラスタリング」の手法がよく用いられます。
階層的クラスタリングでは、sample2 に一番似ているのは、sample3、その次は、sample1 というように、似ている順序が示されます。また、その順序は、系統樹(ツリー)で表現されます。
最終的には、各サンプルのヒートマップの上部(または下部)に、そのツリーを組み合わせてものが結果として用いられます。ツリーとヒートマップは独立していることに留意してください。ヒートマップを書かなくてもツリーは算出できますし、ツリーを算出したからといって、ヒートマップができるわけではありません。(ツリーの結果だけ、ヒートマップに後付けされることもあります。)MeV などのツールを用いれば、クラスタリングの結果が自動的にヒートマップになるので、誤解しやすい点です。
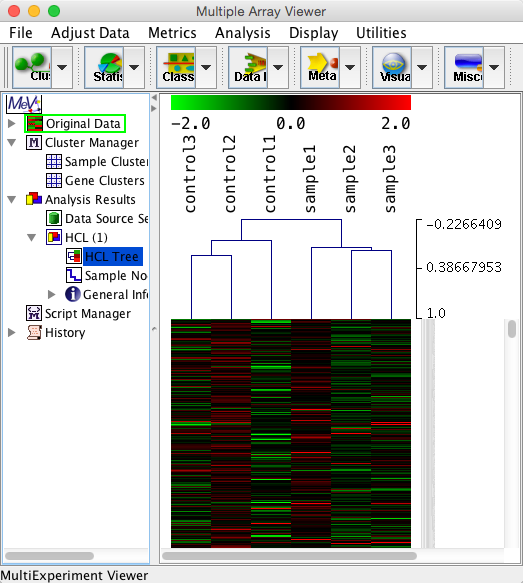
例えば、解析例1のデータをサンプル方向でクラスタリングすると下図のようになります。あえて、遺伝子方向のクラスタリングは行っていません。そのため、ヒートマップの色は分かれているように見えていませんが、どのサンプルが似ているかはツリーから確認できます。
遺伝子の変動パターンを分類するには、クラスタリングが用いられます。MeV を用いて、クラスタリングの手法の1つである k-means を使った例を紹介します。
MeV でデータを読み込み、クラスタリング手法(アルゴリズム)を選択します。(ここでは、クラスタリングの前に、log2変換と中央値からの距離に直す補正を行っています。操作方法はこちら)
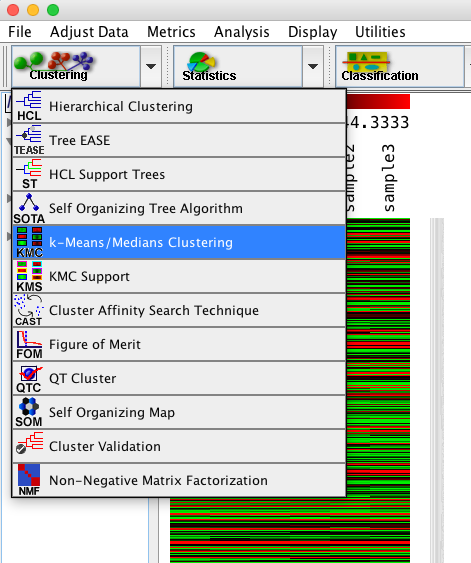
ダイアログが表示されるので、パラメーターを設定します。ここでは、変動パターンで分類したいため、遺伝子方向のみのクラスタリングを行います。(似ているサンプルを探すのなら、サンプル方向にクラスタリングします。)
クラスターをいくつに分けるか指定します。標準では10個の設定です。
また、結果を見やすくするため、さらに階層的クラスタリングで処理するチェックを入れます。
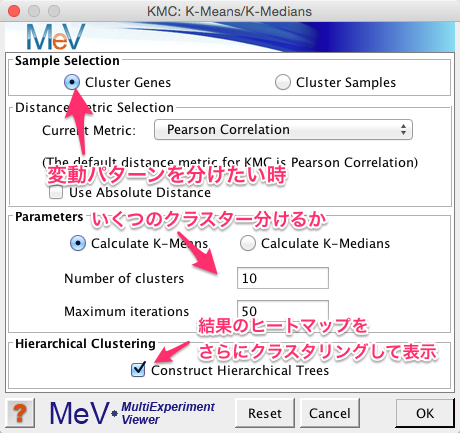
結果の表示用の階層的クラスタリングのパラメーターを設定します。同様に遺伝子方向のみ指定しています。また、最適化のオプションにチェックを入れています。ほかのパラメーターは標準設定のものを使用しました。
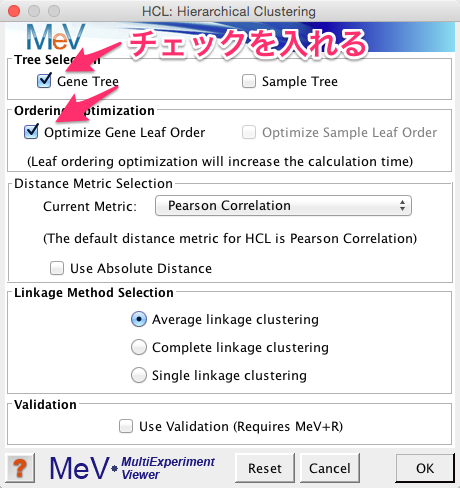
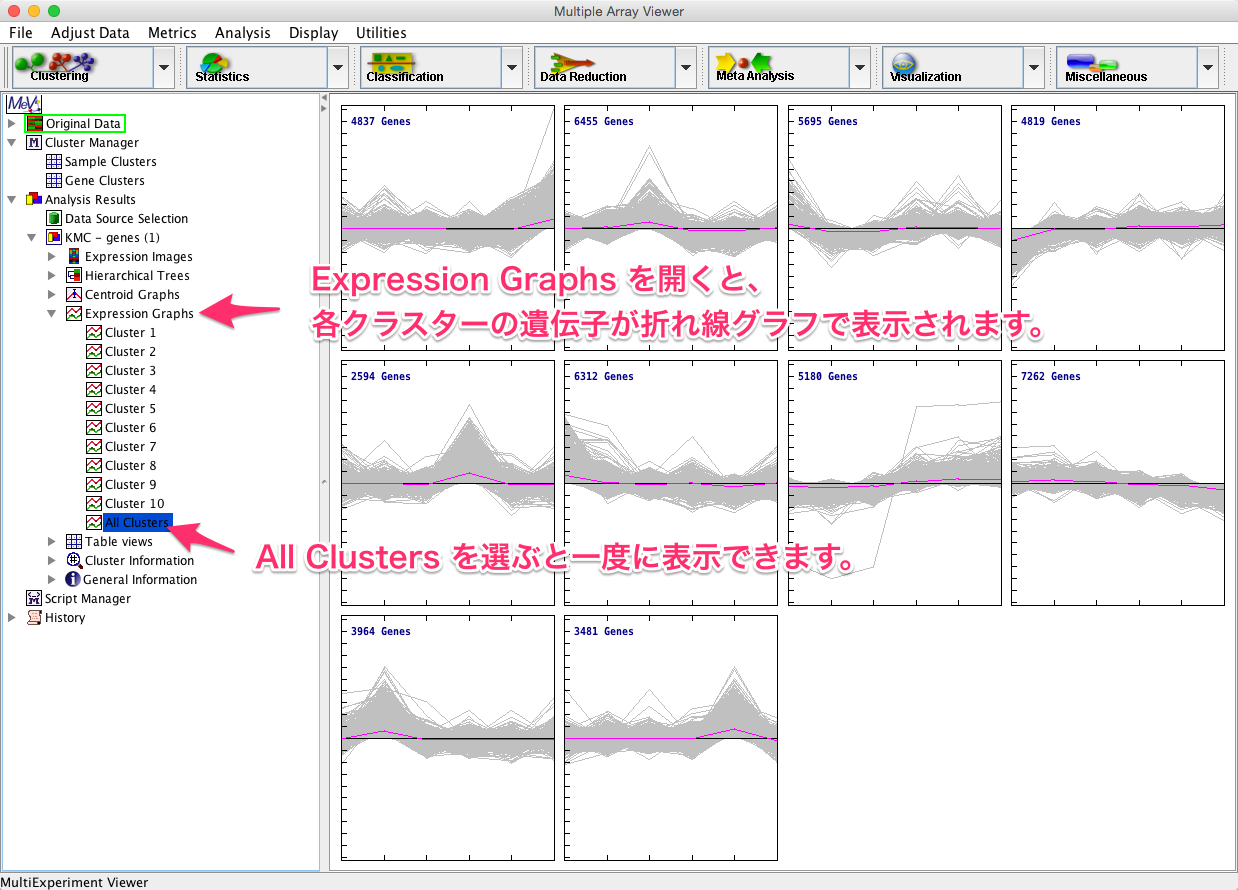
ウィンドウの左側の結果をたどると、変動パターンで10個に分けられた各クラスターのヒートマップを確認できます。
また、 Expression Graphs を選択すると、各クラスターに含まれている遺伝子の折れ線グラフが表示されます。各クラスターに含まれている遺伝子の数もここでチェックできます。
{kind=link}