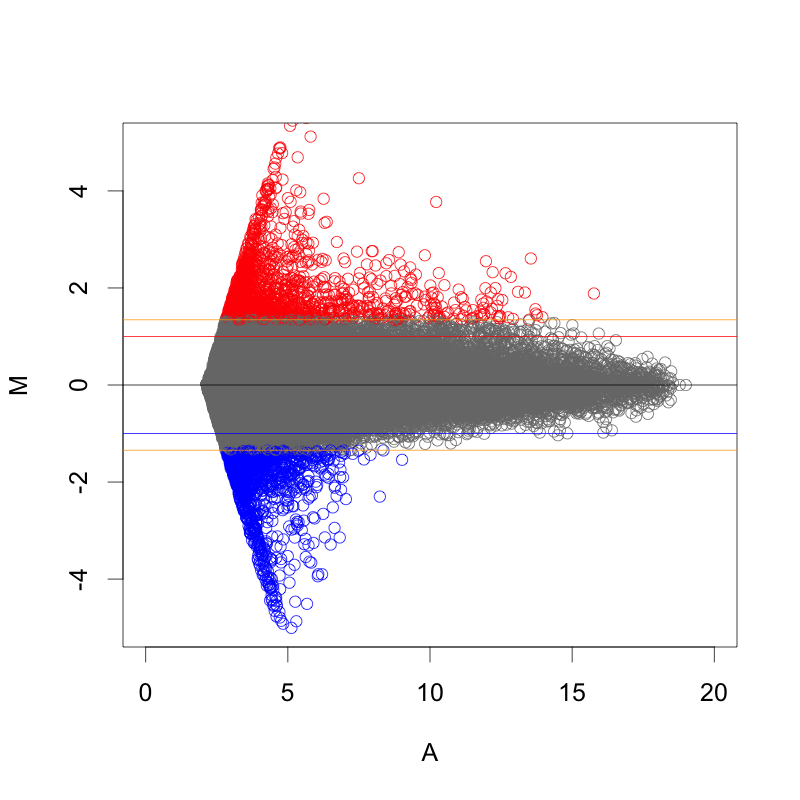
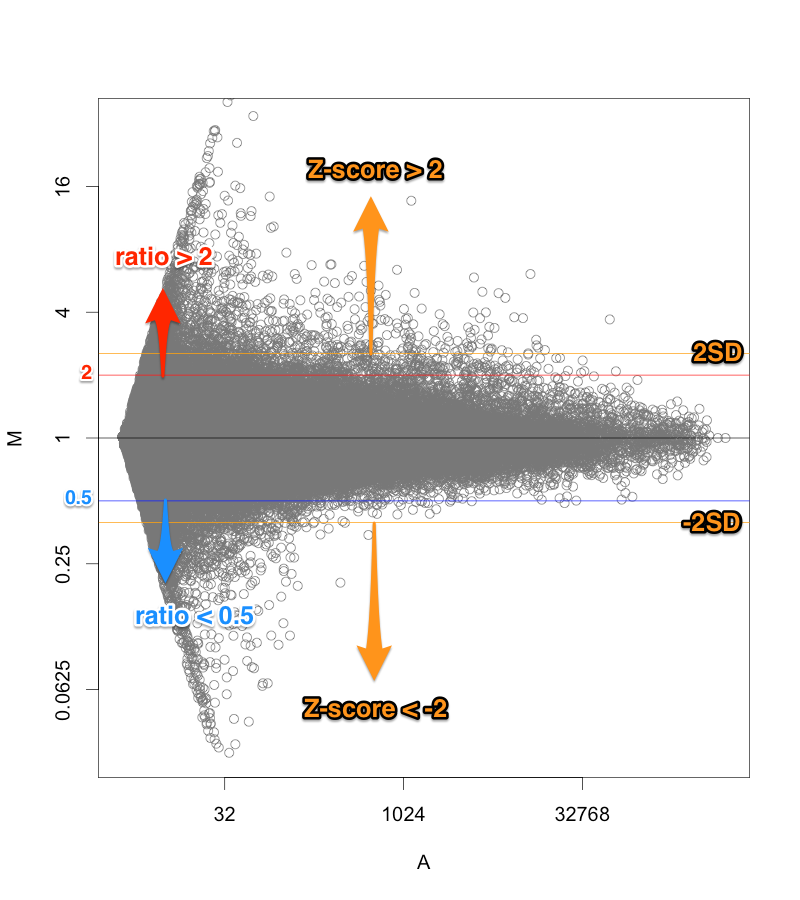
MAプロット で確認すると、 ratio と Z-score の関係が、分かりやすいと思います。解析例1 のデータ (sample2/control2) においては、ratio=2 のライン より、2SDのライン が外側にあります。この場合は、 Z-score のほうが厳しい判定と言えます。(追記:この例の場合、2SD=約1.344 (logFCで)、ratio > 2.54 が Z-score > 2 のラインです。)
MAプロットに示した Z-score の関係。 また、 logFC を Z-score 化して、発現変動遺伝子の判定を行う場合、利点と欠点があります。
Z-score を判定に用いた場合の利点
単純に ratio で判定する場合に比べて、下記のような利点があります。
判定の基準で迷わない。
発現変動が大きいデータの場合、カットオフに使える。
発現変動が小さいデータでも、発現変動遺伝子を判定できる。
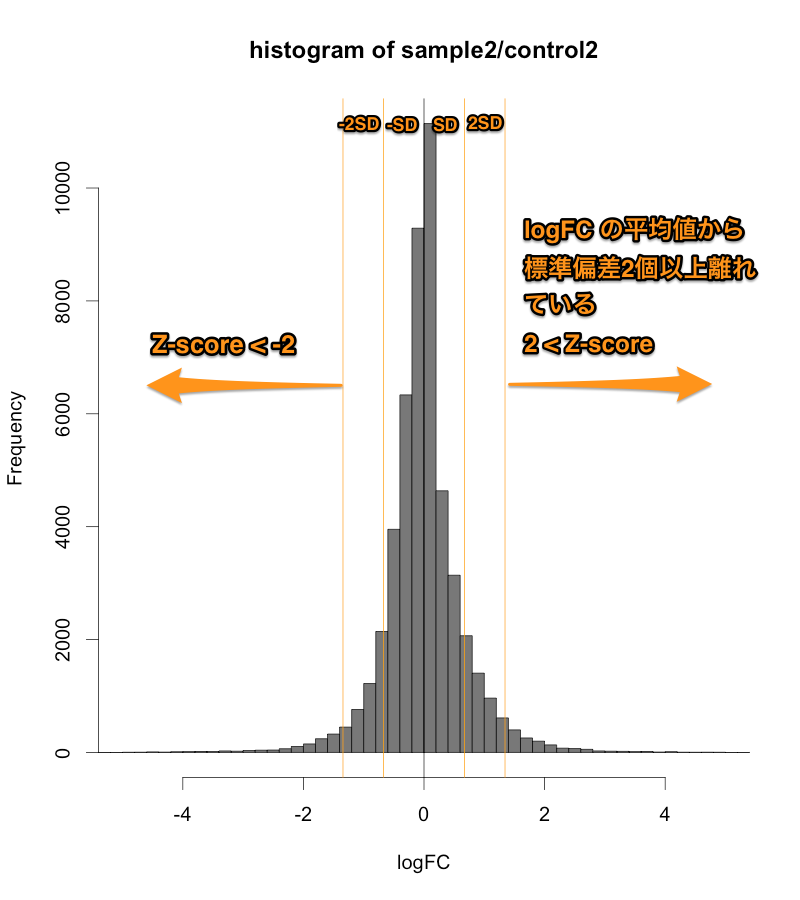
Z-score > 2 または、 Z-score < -2 ということに、統計学的な意味があるので、判定の基準をいくらぐらいにしようかということで迷う必要はありません。(2 以下にあまり意味がありません。)
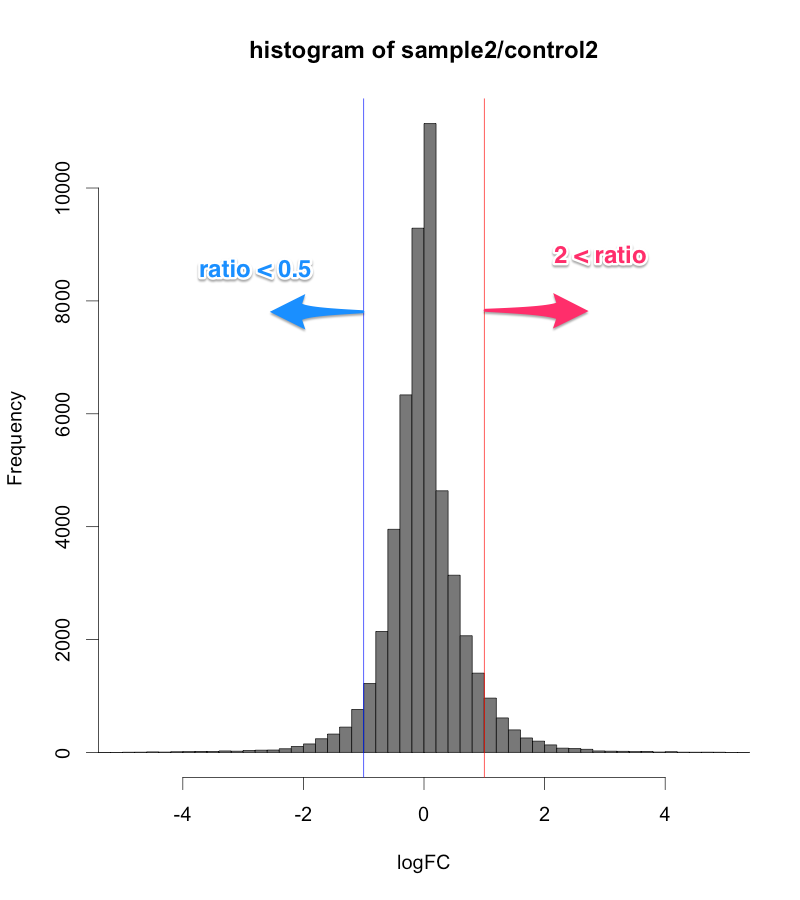
また、発現変動が大きいデータの場合、ratio > 2, ratio < 0.5 などで判定すると、4000個、5000個の遺伝子が、発現変動ありとなることもあります。そのような大きいデータの場合、 Z-score で判定することで、発現変動遺伝子の数を1000個から2000個程度に減らせます。(発現変動遺伝子が多すぎても困ることがあります。例えば、DAVID で1度で解析できる遺伝子の数は、3000個です。)
逆に、サンプルによっては、たかだか1.5倍程度しか変動しないような、小さい発現変動のデータもあります。そのような場合、Z-score だけで判定すれば、(ratioの大きさに関係なく)変動している遺伝子の上位5%程度を取ることになるので、必ず、変動遺伝子を得られます。
Z-score を判定に用いた場合の欠点
利点の裏返しです。
判定が厳しすぎるときがある。
シグナル値が高い部分で、|Z-score| > 2 に比較的なりにくい。
例えば、研究対象としている遺伝子があり、 ratio だと 2 倍を超えているのに、 Z-score だと、 1.7 というようなケースです。Z-score が2以下だからといって、「変動していない」という証明になるわけではありませんが、印象が良くないかもしれません。
また、散布図やMAプロットの先が細いということからも見えることですが、シグナル値の高い部分は、変動が小さく見えがちです。シグナルの高い部分にも、シグナルの低い部分と同程度の標準偏差を求めるならば、高い部分は、|Z-score| > 2 になりにくいといえます。
しかしながら、そもそも、変動が大きすぎたり、変動が小さい場合は、まず、サンプル数(n数)を増やすことを考えるべきです。n=3以上にして、limma, SAM, t-test などの検定を行ったほうが、p-value も算出できますし、より再現性の高い遺伝子を変動ありと判定できます。



{kind=link}