図のタイトル、x軸のラベル、y軸のラベル、凡例 (legend) のタイトルなどの文字は、 labs() 関数で変更できます。
*一方、フォントのサイズや、色の変更は theme() 関数で行います。
labs の変更するオプションとの対応を下図に示します。凡例 (legend) のタイトルの指定は、 fill もしくは、 color で指定します。下記の例は、aes() で、 fill = sample としているため、fill で指定することになります。
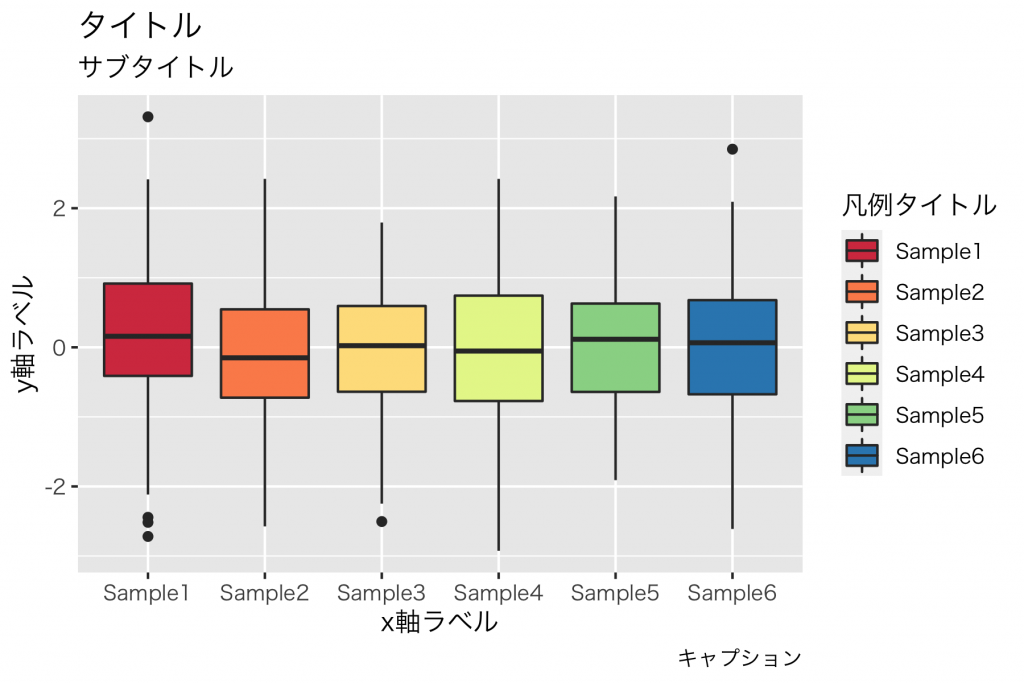
上記のコードの例です。
plot_data <- input_data %>%
gather(starts_with("Sample"), key = "sample", value = "read_count")
g <- ggplot(plot_data, aes(x = sample, y = read_count, fill = sample))
gg <- g + geom_boxplot() +
scale_fill_brewer(palette = "Spectral") +
theme(text = element_text(family = "HiraKakuProN-W3"))
gg + labs(title = "タイトル",
subtitle = "サブタイトル",
x = "x軸ラベル",
y = "y軸ラベル",
caption = "キャプション",
fill = "凡例ラベル")Mac版の R, ggplot2 環境で日本語フォントを使うには、下記を参考にしました。
- https://shohei-doi.github.io/quant_polisci/encoding-r.html
- https://qiita.com/kazutan/items/1f9da9ba2d83db26f3ad