NCBI の GEO にデータを登録する際の注意点など、過去の記事をまとめました。左側のメニューからもたどれます。

NCBI の GEO にデータを登録する際の注意点など、過去の記事をまとめました。左側のメニューからもたどれます。
この解析例の紹介では、遺伝子発現のデータはどういうものか実感できるように、いろいろな種類のデータを取り上げたいと思います。
単純なタイムコースのデータに続き、今回は、iPS細胞のデータを取り扱います。
例のように GEO から、 iPS 細胞のマイクロアレイデータを取得します。GSE42445 のデータです。様々なバリエーションのヒトのiPS細胞 (hiPS) と幹細胞 (hES)、がん細胞 (MCF7など) のデータです。70サンプルあります。
ここでは、rawデータを取得し、 シグナル値を取り出して、 quantile 法で正規化しました。(正規化後のデータ: Series Matrix File を取得して、そのまま用いても良いでしょう。)
正規化前 (raw) のデータをボックスプロットで表示すると下図のようになります。特に大きく外れたサンプルはないようです。右端のがん細胞のデータが若干低いようですが、よくある程度だと思います。
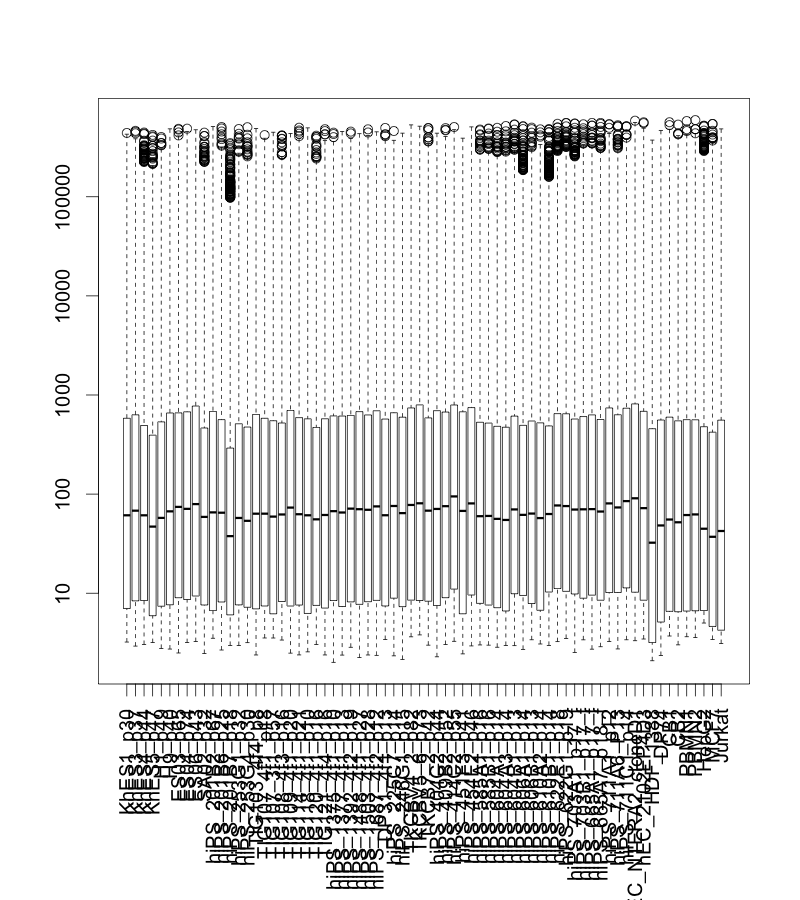
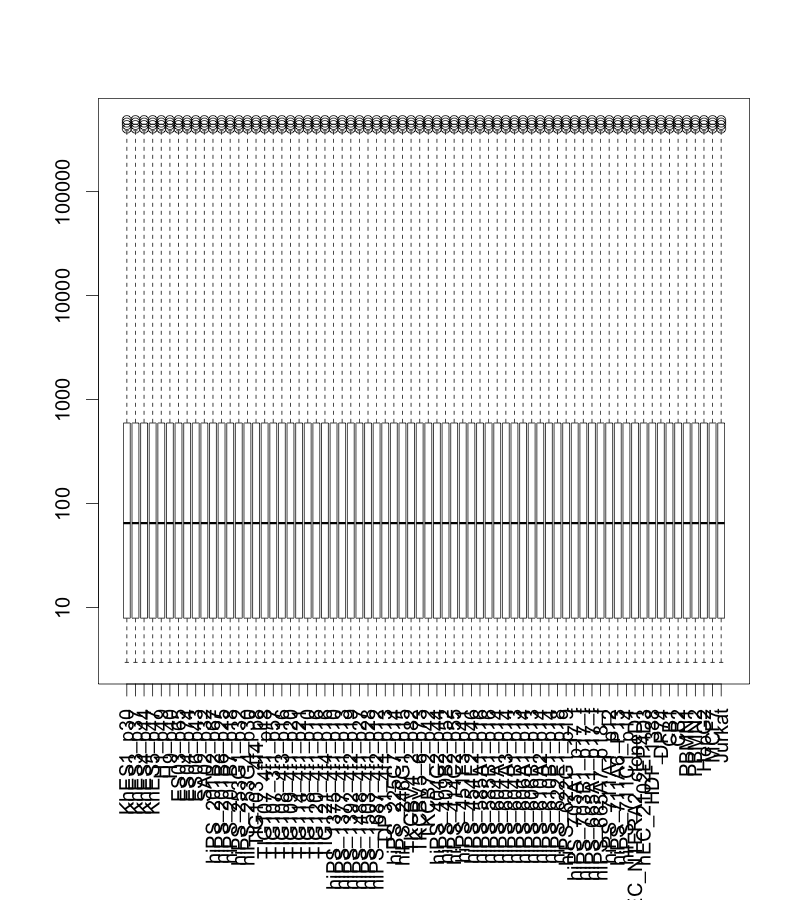
正規化後のデータをボックスプロットで表示すると下図のようになります。データの分布がそろっていることが確認できます。
ヒートマップとクラスタリングを応用することで、特定の組織で発現の高い遺伝子を探すことも可能です。
例えば、ES細胞だけで発現の高い遺伝子を探してみましょう。
まず、データとしては、様々な組織のマイクロアレイデータが必要になります。個々の研究室レベルで揃えようと思うと大変ですが、幸い近年では公開データがあるので、うまく利用しましょう。
いろいろな組織の遺伝子発現を比較したければ、 BioGPS のデータが使えます。心臓、肝臓、肺などの種々の組織、約180サンプルぶんのマイクロアレイデータがあります。
データを取得したら、正規化して、クラスタリングを行い、結果をヒートマップで表示します。ここでは、RMA と quantile アルゴリズムによって正規化し、階層的クラスタリングを行いました。
全遺伝子では結果を見るのが大変なので、正規化後、アノテーションを元に stem cell 関連遺伝子だけを抽出しています。また、MeV上で、シグナル値を log2変換して、中央値からの距離に変換しています。クラスタリングは、遺伝子方向、サンプル方向とも階層的クラスタリングで行っています。
結果は下図のようになります。遺伝子が縦に並んでいます(行)。組織(サンプル)が横に並んでいます(列)。
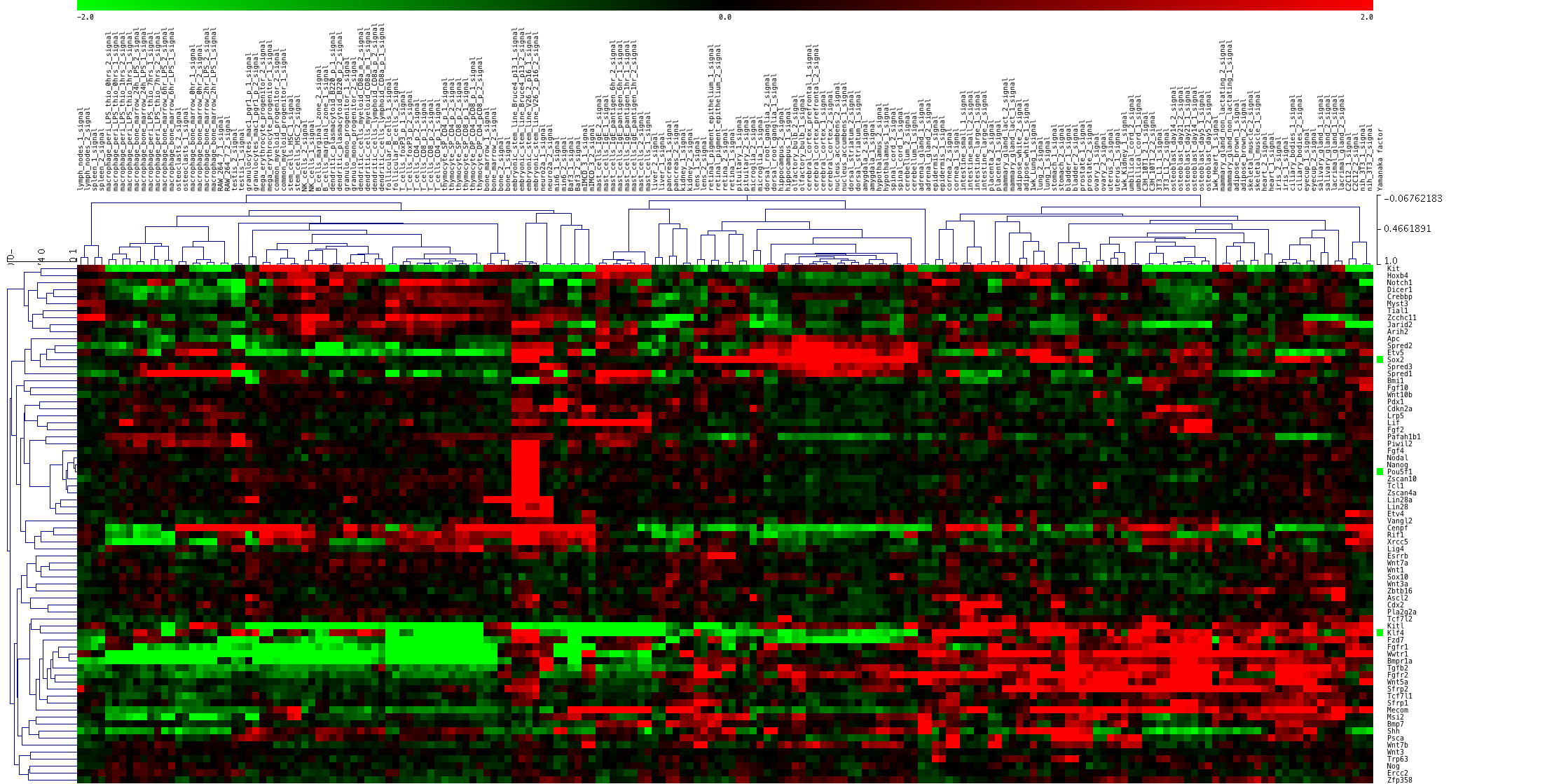
左から3分の1ほどの列に、ES細胞のデータ(4列)が並んでいます。これらの4列だけで、赤くなっている遺伝子があれば、それらが、ES細胞だけで発現が高い遺伝子です。
探してみると、真ん中より少し上にある遺伝子のクラスターに、ちょうど4列だけ赤い部分があります。ハイライトすると下図の部分です。遺伝子名を確認すると、Pou5f1 = Oct3, Nanog など有名な遺伝子が該当しているのが分かります。(見やすいように横方向を縮小しています。)
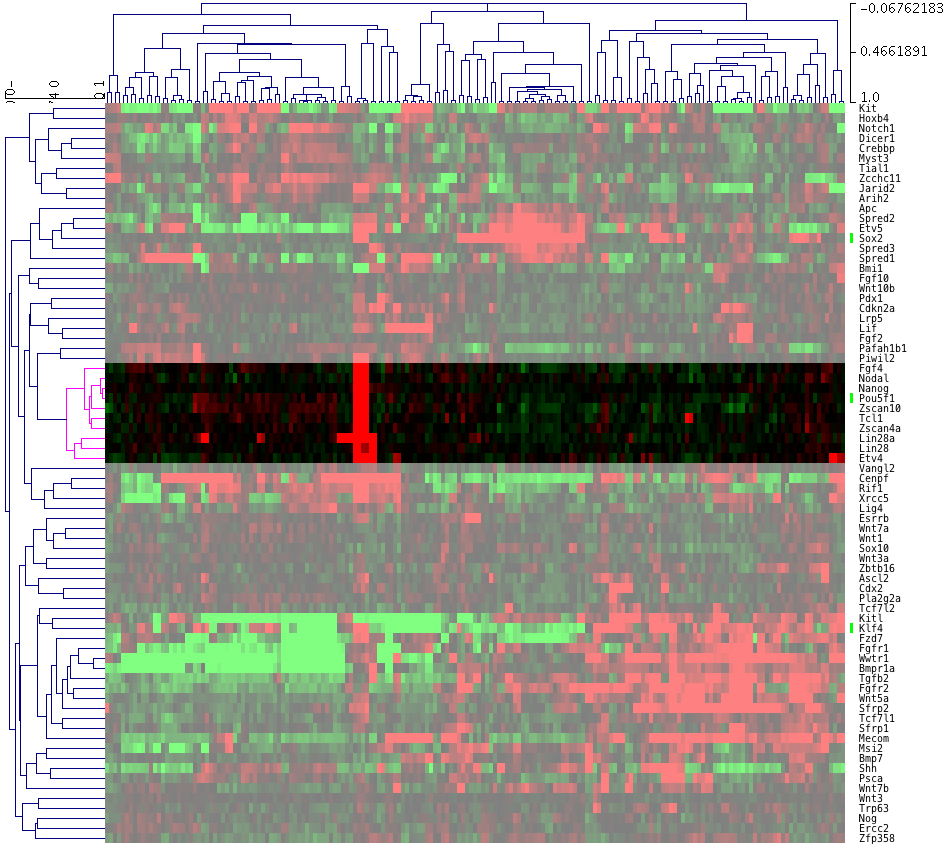
なお、色付けの設定から、「黒=発現していない」ではない、ので注意してください。
ボックスプロットを確認したら、次は散布図 (scatter plot) も確認してみましょう。
例として、 control2 と sample2 を比較した場合の散布図を示します。 ratio > 2 のプローブ(=遺伝子)を赤、 ratio < 0.5 の遺伝子を青に色づけしています。正規化後データを用いています。
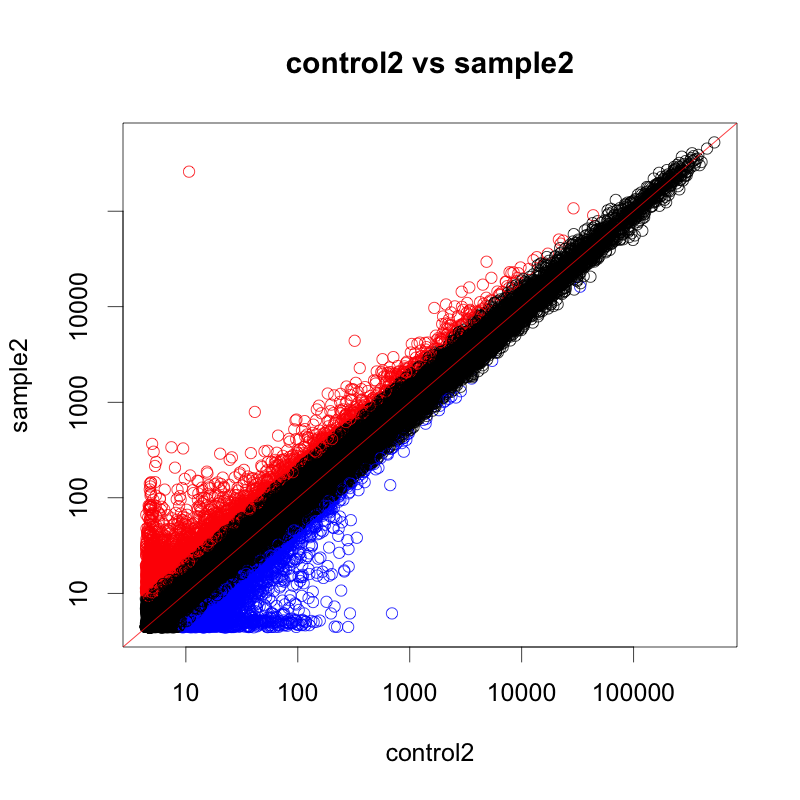
散布図の広がり方から、平均的なデータのように見えます。(がんのサンプルや、変動が大きいデータでは、もっと点が全体に散らばって見えます。)
rawとして、正規化前のデータの散布図も作成しました。

よく見ると、rawデータと、正規化後のデータでは、(ratio で判定した場合)変動ありと判定される遺伝子に異なる部分があることがわかります。特にシグナル値の高い部分です。
散布図の左下から右上に引かれた赤線は、 y=x を意味しています。raw データの散布図は、集団が y=x より下に膨らんで見えています。そのため、rawデータをそのまま用いると、sample2で減少した遺伝子が多く見つかり、増加した遺伝子は少なく見つかることになります。
一方、正規化後データの散布図では、点の中心が y=x 上に載っていることが分かります。(=ほとんどの遺伝子が変動していない。偏りがない。)
このように散布図を確認すると、raw, 正規化前のデータに偏り(バイアス)がないか、また、正規化後のデータから偏りが解消されているのか確認できます。
スコアだけで判断して、結果を誤って解釈しないよう、散布図を必ずチェックするようにしましょう。
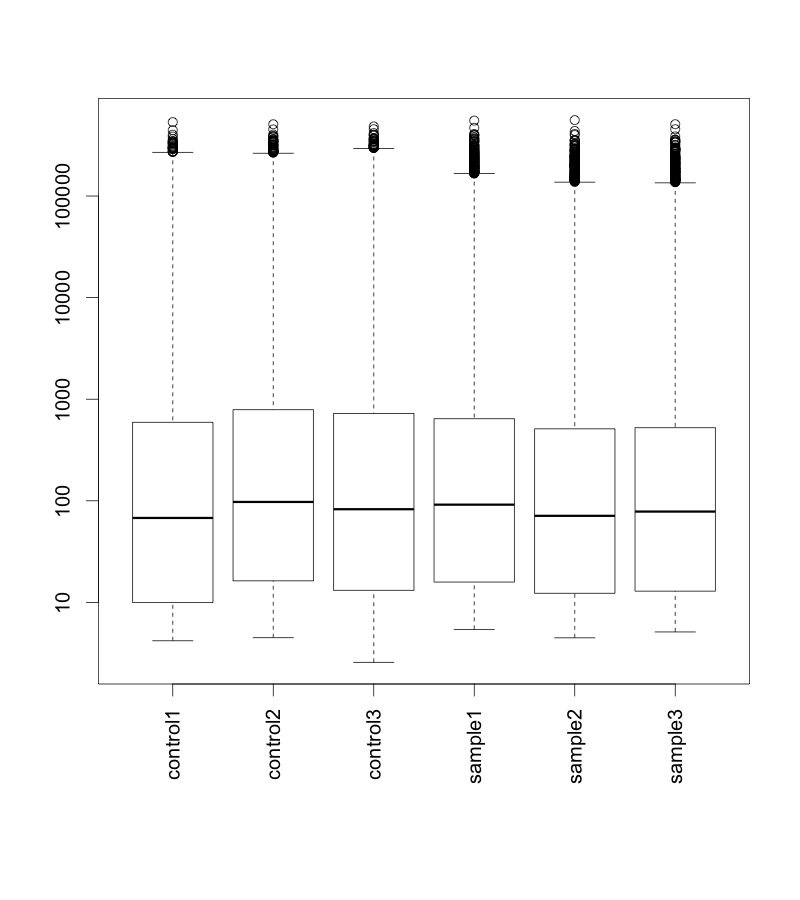
ダウンロードしたデータを用いて、ボックスプロットを作成してみましょう。マイクロアレイデータは、まず、ボックスプロットや散布図を書いて、シグナル値の状態を確認することをおすすめします。
シグナル値のばらつきが極端に大きい場合は、データのクオリティが良くない(サンプルのコンディションが悪い、RNAの分解が進んでいる)ことも考えられれます。
raw データ(正規化前)の散布図を示します。便宜上、サンプルの名前を、293T_16hr_Control = control1, 293T_16hr_muTRPV3 = sample1 のように変更しています。サンプルによって、多少上下していることが確認できます。サンプルのクオリティは悪くないように見えます。
使用した raw データの値は、下記よりダウンロードできます。
> https://www.dropbox.com/s/igxolub38mrm0po/rawdata.txt.zip?dl=0
(Agilent の raw データのファイルからシグナル値だけを取り出す方法については割愛します。)
続いて、正規化後データ (normalized data) のボックスプロットです。ここでは、正規化のアルゴリズムとして、 quantile 法* を用いています。データの分布がそろっていることが確認できます。

使用した正規化後データの値は、下記よりダウンロードできます。
> https://www.dropbox.com/s/2m1poolbqz9vizh/normalized_data.txt.zip?dl=0
極端に分布の異なるサンプルは、正規化の際(アルゴリズムによっては)、他のサンプルのシグナル値にも影響を与えることもあります。物理的なサンプルの状況を確認して、RNAの分解などが疑われる場合は、そのサンプルを除外して正規化を行ったほうがよいでしょう。
* Bolstad et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics (2003) vol. 19 (2) pp. 185-93.