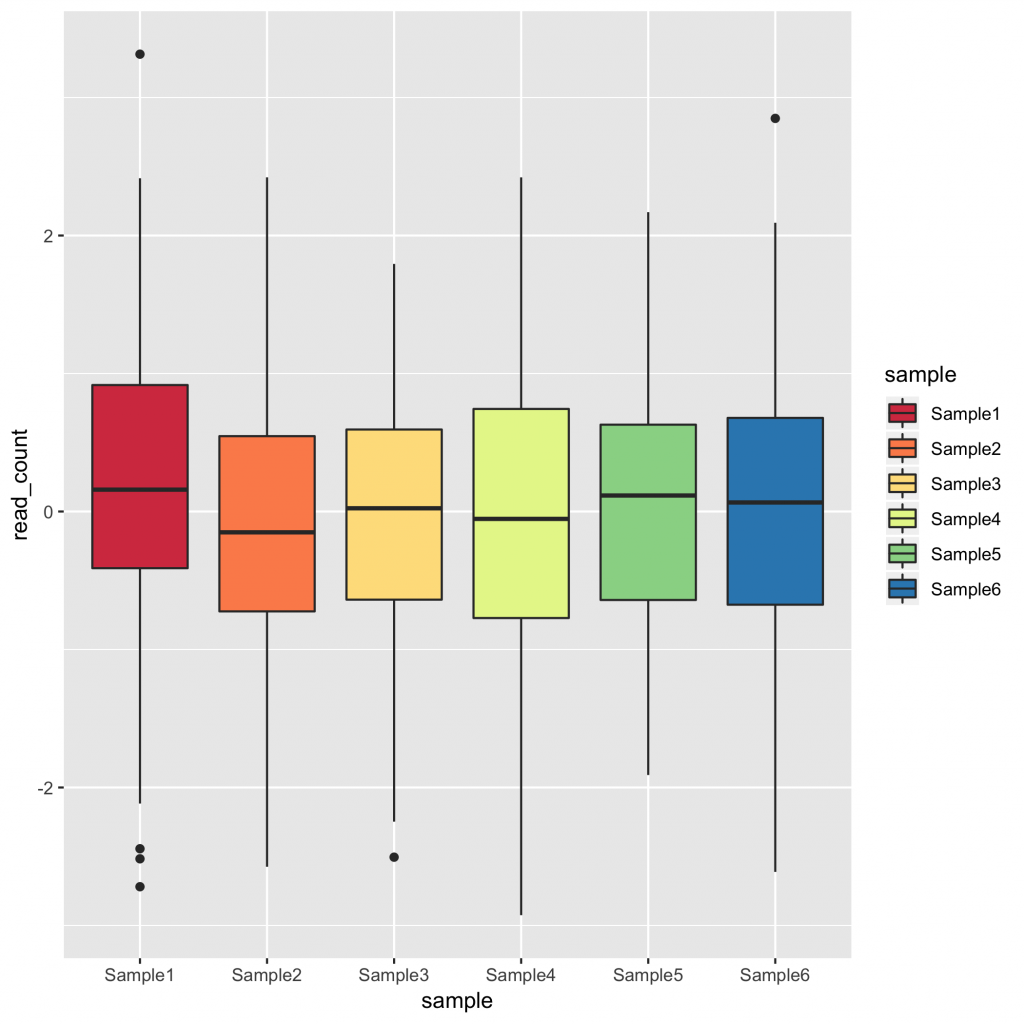
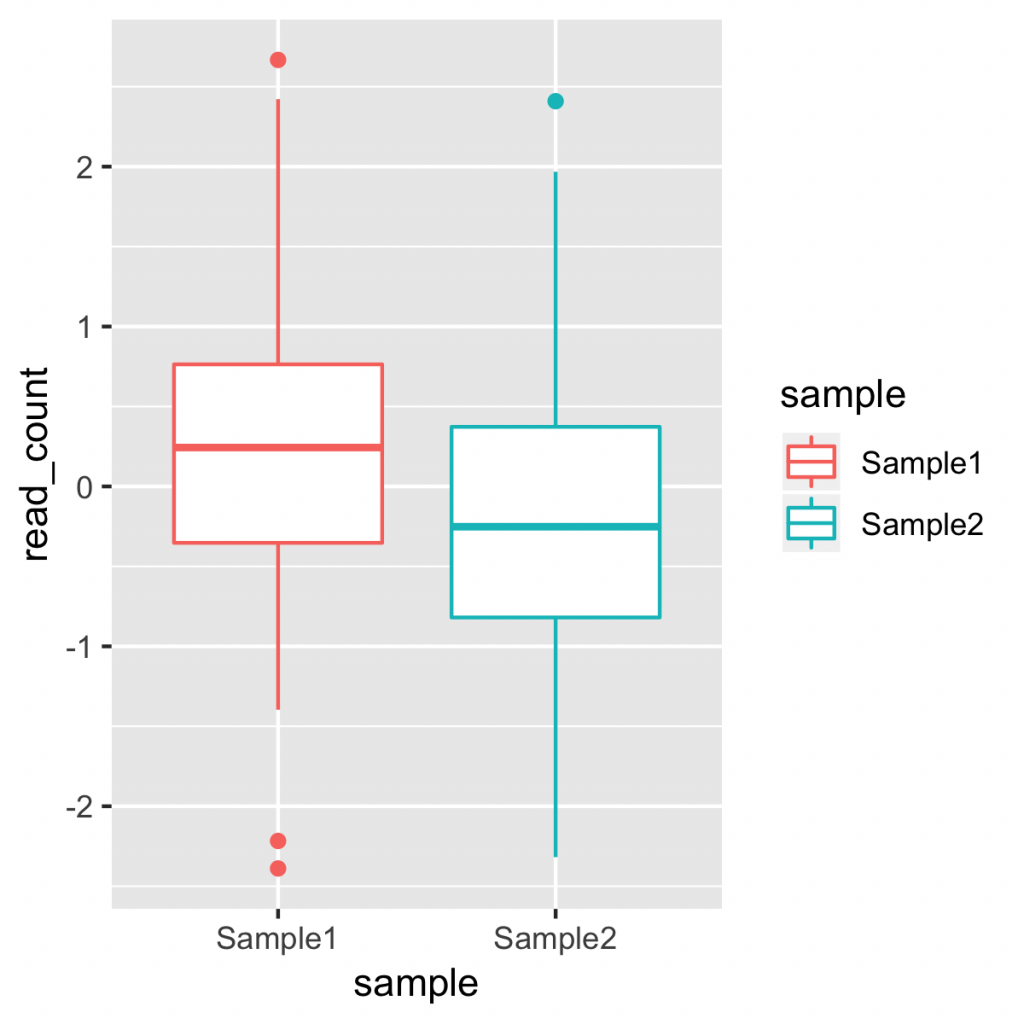
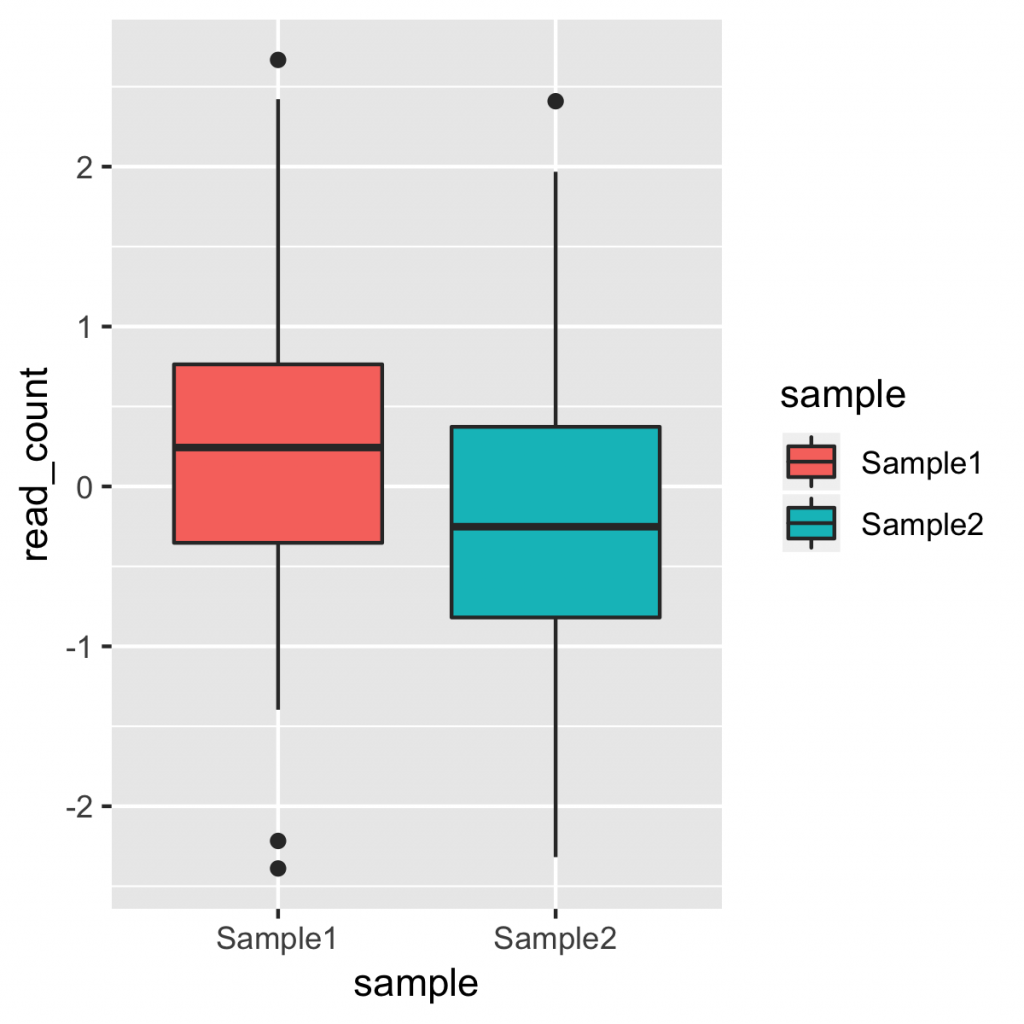
全体のフォントのサイズを変更するには、 theme() 関数を用います。オプションの「text」にフォントのサイズを指定します。その際、直接、「size = 24」 とするだけではなく、 element_text() の中で宣言する必要があります。
theme(text = element_text(size = 24))
theme() 関数は、geom_boxplot() の後に 「+」で続けて指定します。
g <- ggplot(plot_data, aes(x = sample, y = read_count, fill = sample))
g + geom_boxplot() + theme(text = element_text(size = 24))
ggsave("boxplot_image.png", width = 4, height = 4, unit = "in")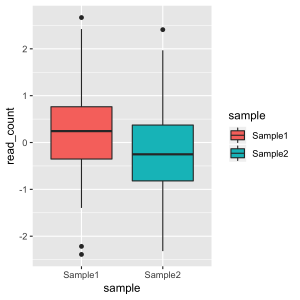
theme 関数の text オプションは、 x 軸や y 軸のラベル、キャプションなど、全体に影響します。個別に指定する場合は、 theme(axis.text.x = element_text(size = 24)) などとします。
その他のテキストについても、個別に細かく指定が可能です。
- x軸のラベル: axis.text.x
- y軸のラベル: axis.text.y
- x軸のタイトル: axis.title.x
- y軸のタイトル: axis.title.y
- 凡例のタイトル: legend.title
- など、他にもあります。
Modify components of a theme
https://ggplot2.tidyverse.org/reference/theme.html