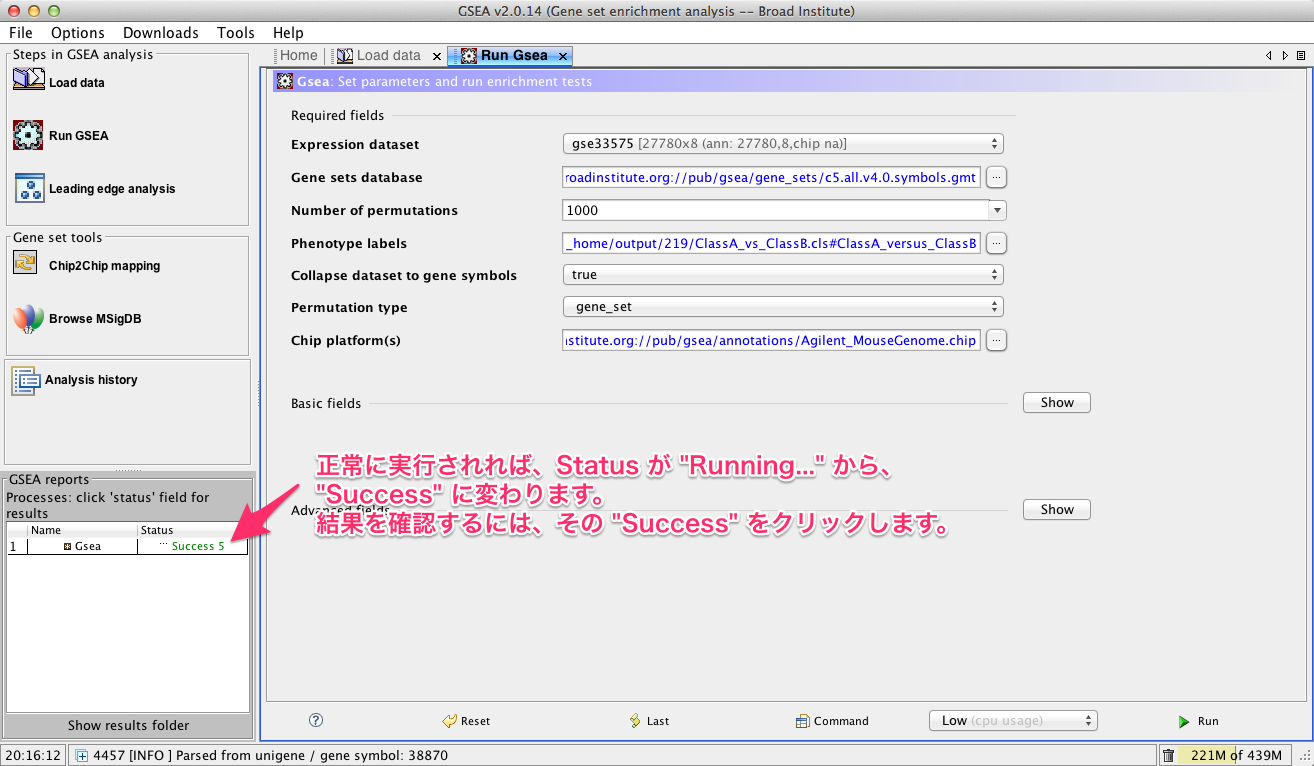
GO解析の考え方
発現が増加または、減少した遺伝子の一群(発現変動遺伝子群)の中に、「特定の用語(機能、キーワード)をアノテーションに持つ遺伝子が多い」ことを述べるには、下記の2つの点を考慮する必要があります。
- その用語をアノテーションに持つ遺伝子が、ゲノム中にもともと多いのかどうか。
- 発現が増加または、減少した遺伝子の数が多いかのどうか。
1. について
もともとゲノム中に多く含まれる機能は、当然、発現変動遺伝子群においても見つかりやすいです。たとえば、 GO:0005488 : binding は、ほとんどの遺伝子がこの機能をアノテーションされています。(何らかのタンパクと結合することが考えられますので、当然といえます。)GO:0016020 : membrane などもそうです。ほとんどの遺伝子は、膜系か、それ以外に分類されるためです。
「当たり」が多く含まれている宝くじを引いていることをイメージするとよいでしょう。それでは当たったことが重要にはなりません。
2. について
発現が増加または、減少した遺伝子の数が多い場合、あらゆる機能の遺伝子が見つかりやすくなります。マイクロアレイデータによっては、3000個以上の遺伝子が増加、減少していることもあります。この状態では、ほぼすべての機能の遺伝子が含まれていて当然と考えられます。
宝くじをたくさん引いていることをイメージするとよいでしょう。何度も挑戦することで、当然、当たりやすくなるため、当たったことの重要性は薄れます。
好ましい状態
上記を考慮すると、最も重要性が高くなるのは、次の条件です。
「もともとゲノム中に数少なく見られる機能を持つ遺伝子が、少ない発現変動遺伝子群に数多く含まれている。」
言い換えると、「ゲノム中(4万個)に10個程度しか含まれない遺伝子が、マイクロアレイ解析の結果、変動していた100個の遺伝子群の中に、8個も見つかった」というような状況です。(数字は適当です。)
DAVID の解析結果などに表示されている GO についた p-value や Enrichment Score は、上記の点を考慮して算出されたものです。 p-value < 0.05 であれば、偶然ではないことが主張できます。