MeV を用いて、ペアを考慮した t-検定を行う方法を紹介します。
検定を行う2グループに含まれるサンプルの間に、ペアの関係がある場合は、ペアを考慮した検定が可能です。(例えば、患者ごとに投与前、投与後のサンプルある場合など。)ここでは、仮に WT と KO にペアの関係があったと仮定して、例として用いています。
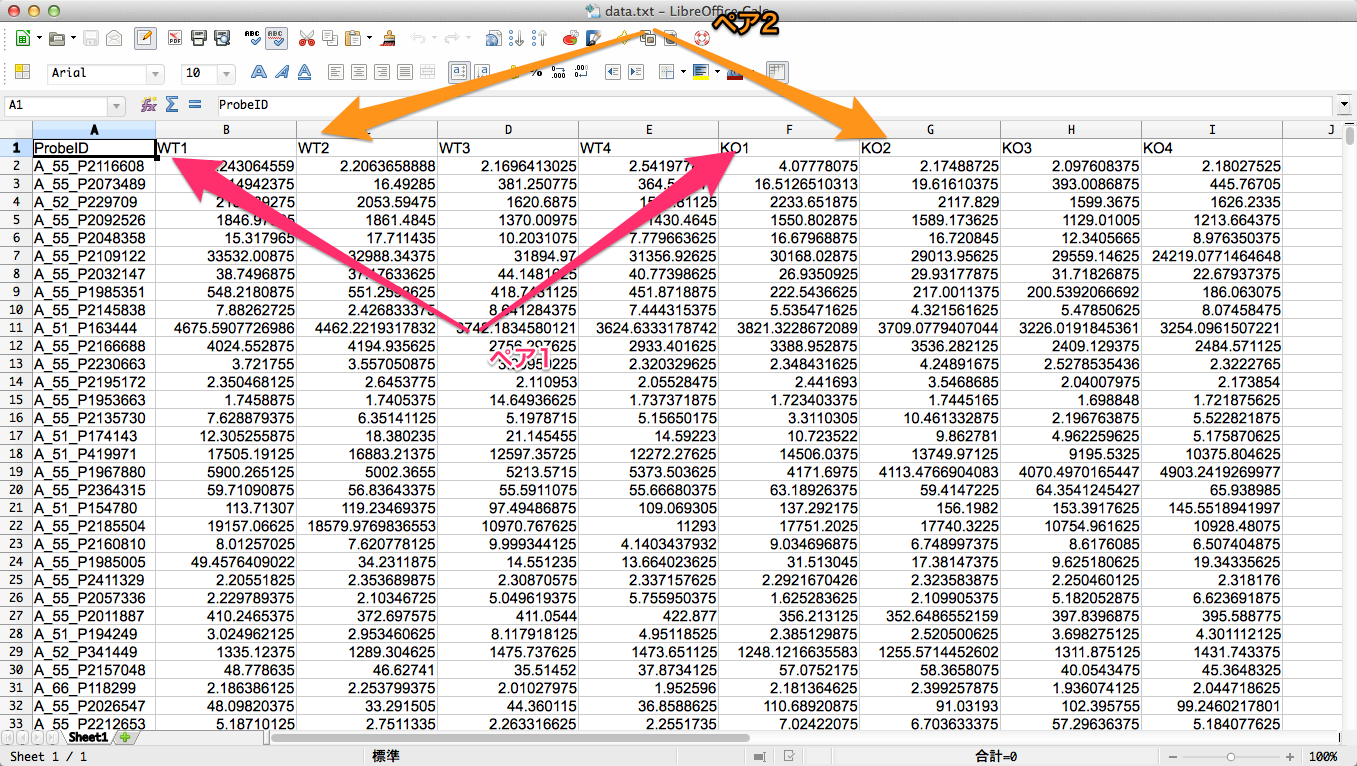
1. データの読み込み
通常通り、データを読み込みます。データが表示されます。
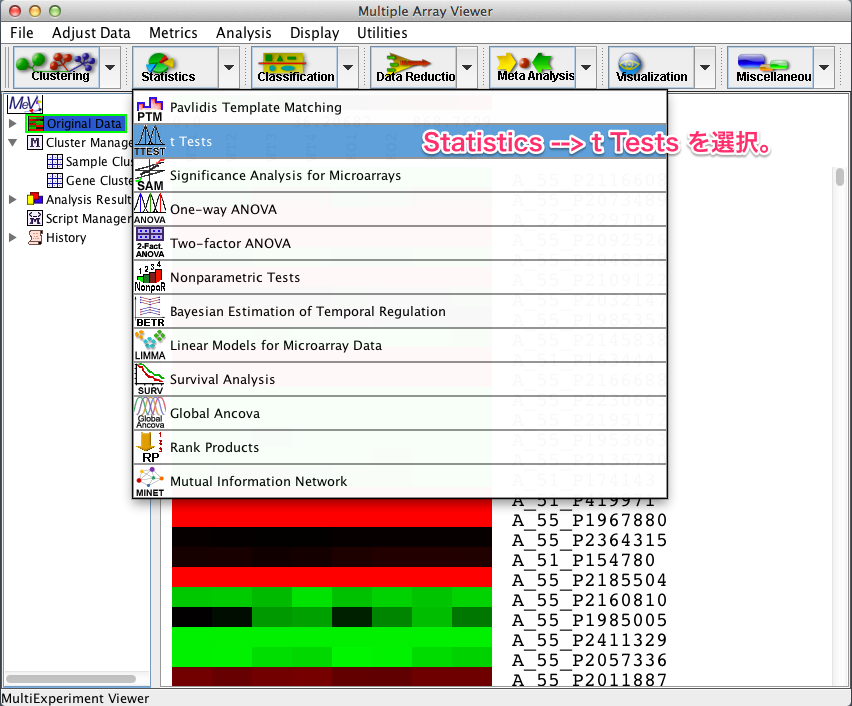
2. t-検定
上部メニューから、 “Statistics” –> “t Tests” を選択します。
2.1 ペアの設定
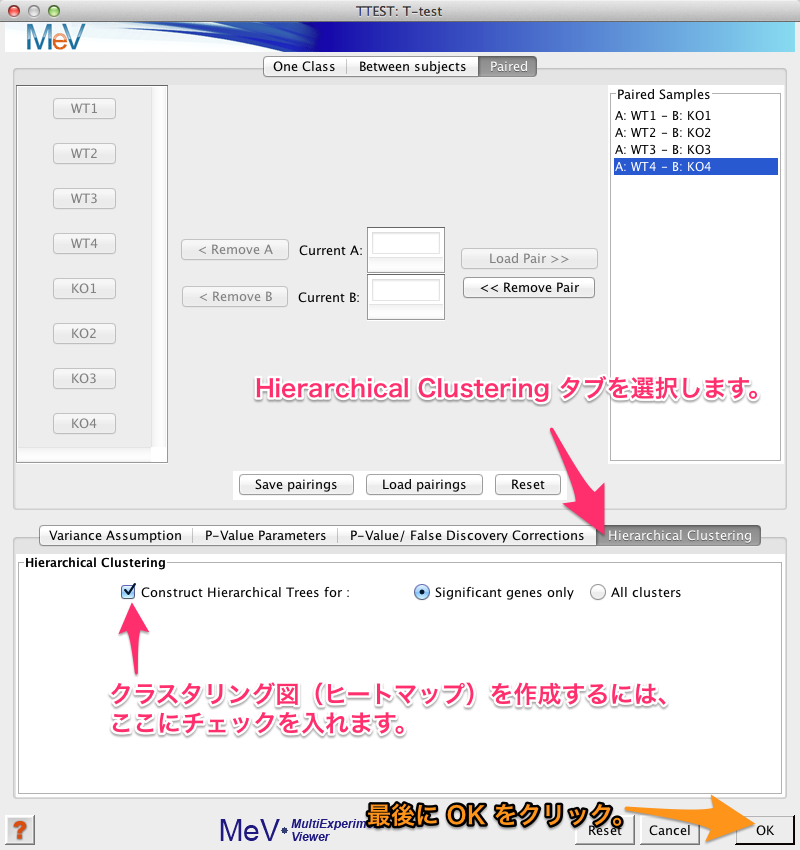
検定のパラメーターを設定するためのウィンドウが表示されます。このウィンドウの上部のタブから、“Paired” を選択します。
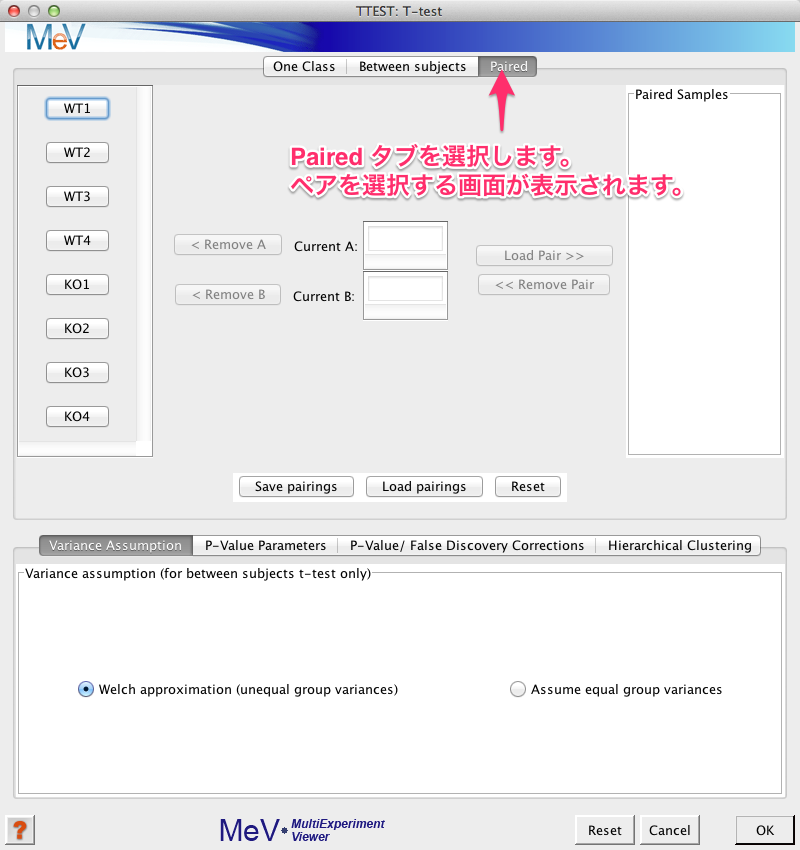
ペアを選択する画面になったら、左側のサンプル名からペアにするサンプルを選びます。
- (1) まず、1つめのサンプル (WT1)をクリックします。
- (2) 次に対応するサンプル (KO1) をクリックします。
- (3)そして、ペアが選択されたことを確認して、 Load Pair を選びます。

ペアが表示されます。
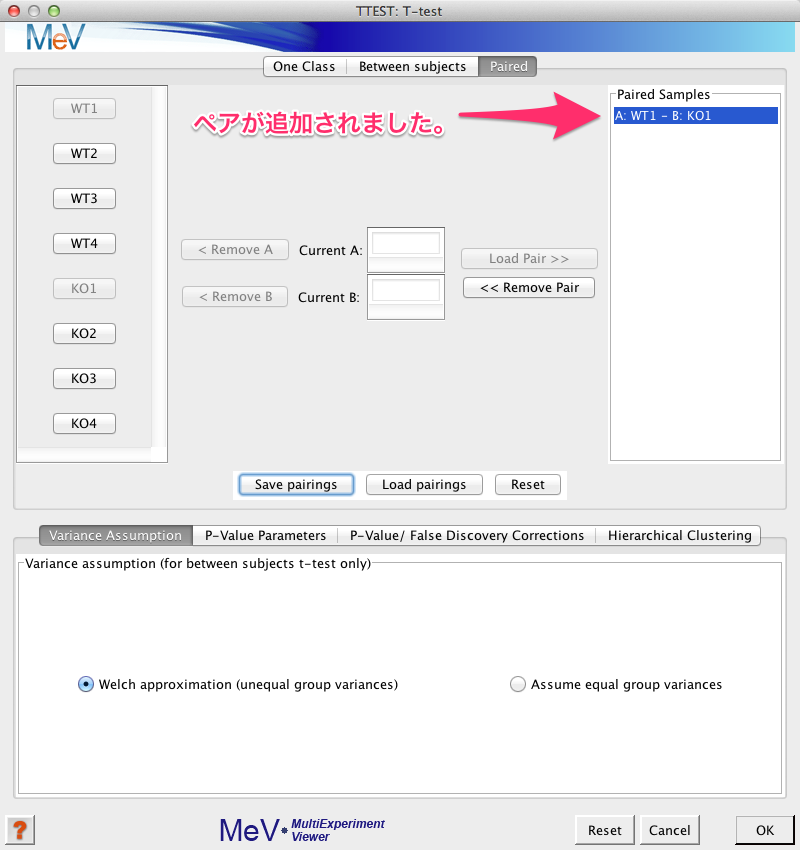
上記の (1) から (3) の動作をペアの数だけ、繰り返します。
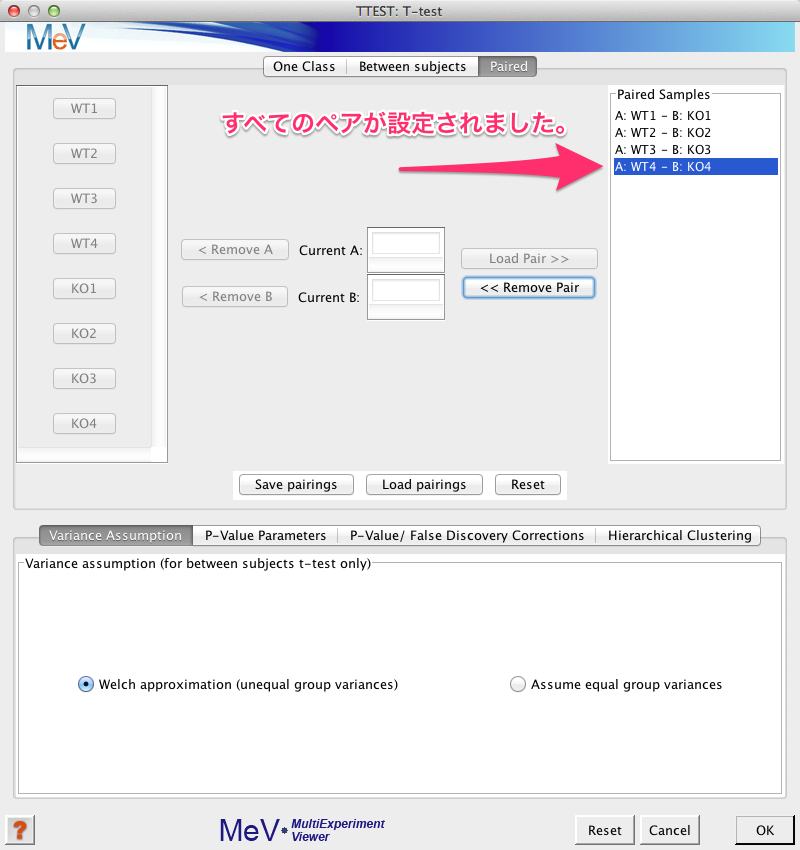
2.2 検定のパラメーターの設定
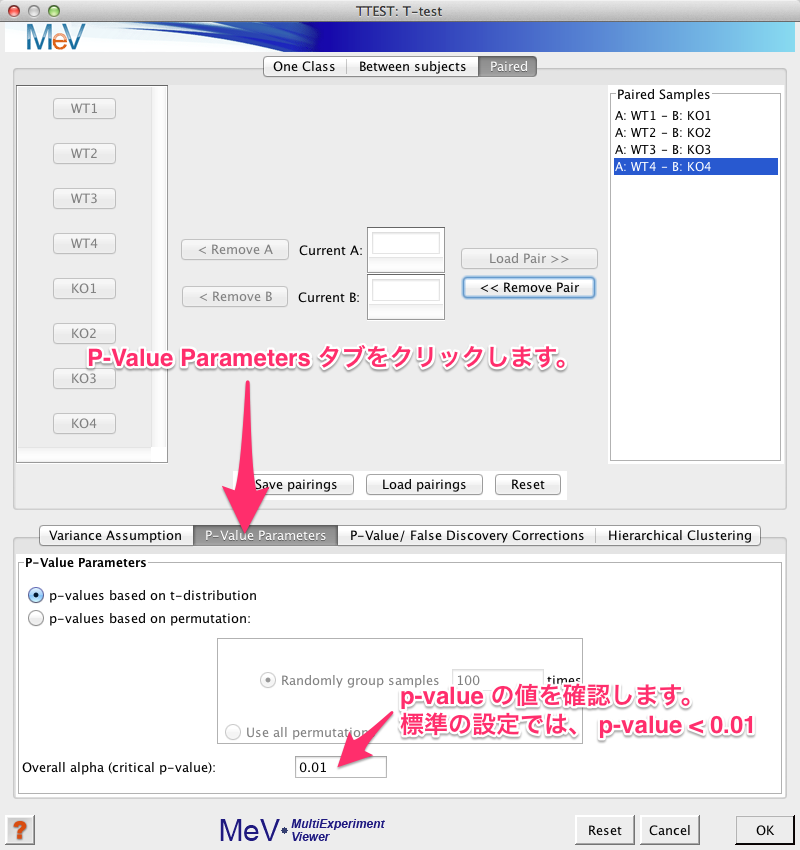
すべてのペアの設定が終わると、検定のパラメーターを設定します。ウィンドの中ほどの “P-Value Parameters” タブをクリックし、 p-value の設定を確認します。標準の設定では、 p-value < 0.01 の遺伝子が有意 (significant) と判定されます。変更の必要があれば、 0.05 などに変更します。
次に、クラスタリングのオプションを設定します。検定結果をクラスタリングされたヒートマップで表示したい場合は、 “Hierarchical Clustering” タブをクリックして、チェックを入れます。
すべての設定が終わったら、最後に右下の OK をクリックします。
2.3 検定結果の表示
OKをクリックした後、クラスタリングの設定画面が表示されますので、オプションを選択します。検定終了後、クラスタリング図などは、左側のツリーに表示されます。三角マークをクリックして、ツリーを開くと、それぞれのヒートマップを確認できます。なお、ヒートマップは、以前に紹介したように適宜、色づけを行ってください。
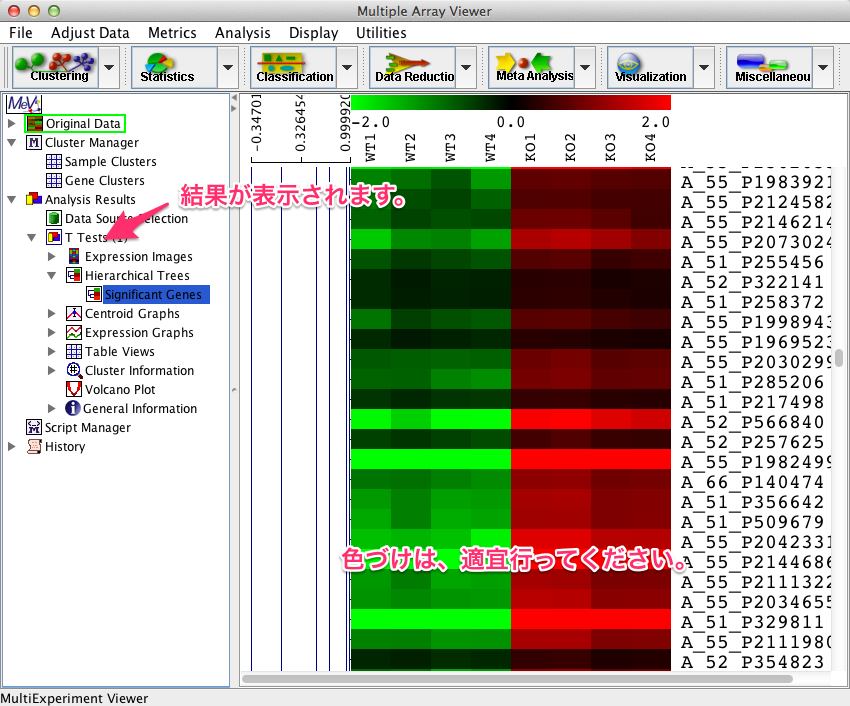
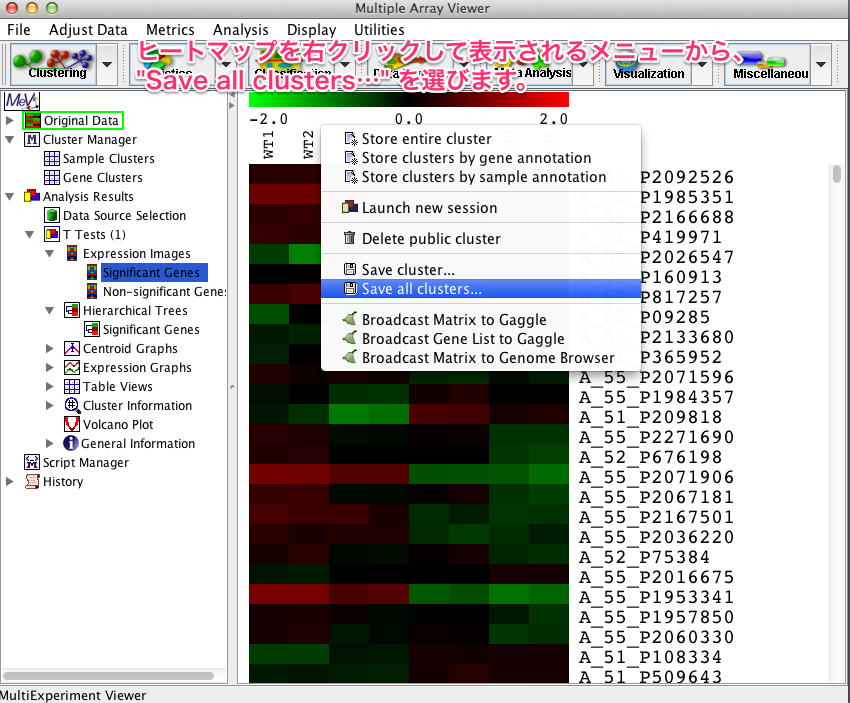
ヒートマップを右クリックして表示するメニューから、 “Save all clusters…” を選ぶと、p-value などスコアを保存できます。