前回は、先に特定の機能の遺伝子をピックアップし、その後、ヒートマップで変動パターンをチェックする方法を紹介しました。これに対し、先に変動パターンをチェックするという方法もあります。
変動パターンで抽出したのち、ヒートマップを確認する
解析例1のデータは、各タイムポイントごとに、3つの比較を行いました。
- 16hr: sample1/control1
- 24hr: sample2/control2
- 40hr: sample3/control3
この3つの比較のいずれかの比較において、変動している遺伝子(増加:ratio>2 かつ Z-score > 2、減少:ratio < 0.5 かつ Z-score < -2)の遺伝子を抽出して、ヒートマップを作成しました。ヒートマップの色付けなどは、これまでと同様です(logFC の中央値からの距離)。図には、2678個の変動遺伝子が含まれています。縮小しているため、画像のサイズは前回のアポトーシスのヒートマップと同じですが、こちらが多くの遺伝子が含まれています。
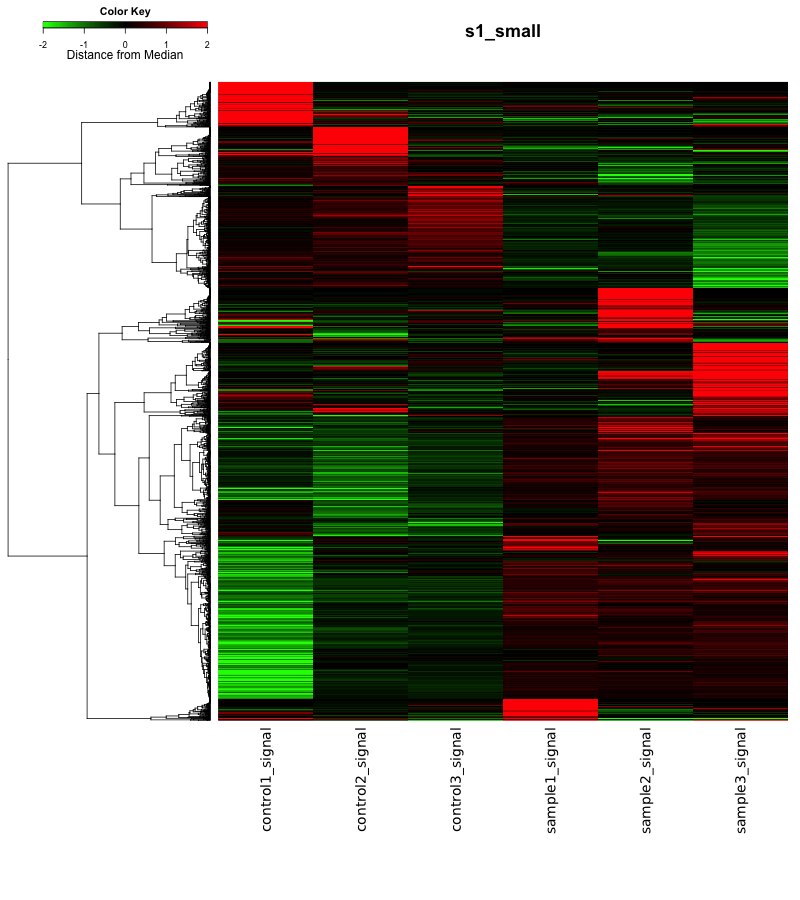
ヒートマップを見ることで、変動パターンを把握できます。例えば、16hrと24hrで共通に増加した遺伝子があるのか、24hrと40hrで共通に減少した遺伝子があるのか、あるとしたら何個くらいか、すべてヒートマップから見て取れます(数値は、おおよそではありますが)。
「共通の変動遺伝子が何個ある」というのをチェックするために、「ベン図」が用いられることもありますが、比較の組み合わせが、2つのときはよくても、今回のように3つ以上の場合は、複雑になりますので、ヒートマップで確認することをお勧めします。(また、マイクロアレイデータの場合、データの見せ方にもよりますが、変動遺伝子の個数には、あまり意味がない場合が多いです。)
特徴的な変動パターンから機能解析 (DAVID)
実験条件から、いくつかの変動パターンが予想されるかもしれませんが、もし、特徴的なパターンがあるのなら、ヒートマップで見えるはずです。このデータの場合、24hrだけで増加、40hrだけで増加という部分が特徴的なようです。(=なぜか、16hrだけ増加は少ない?)
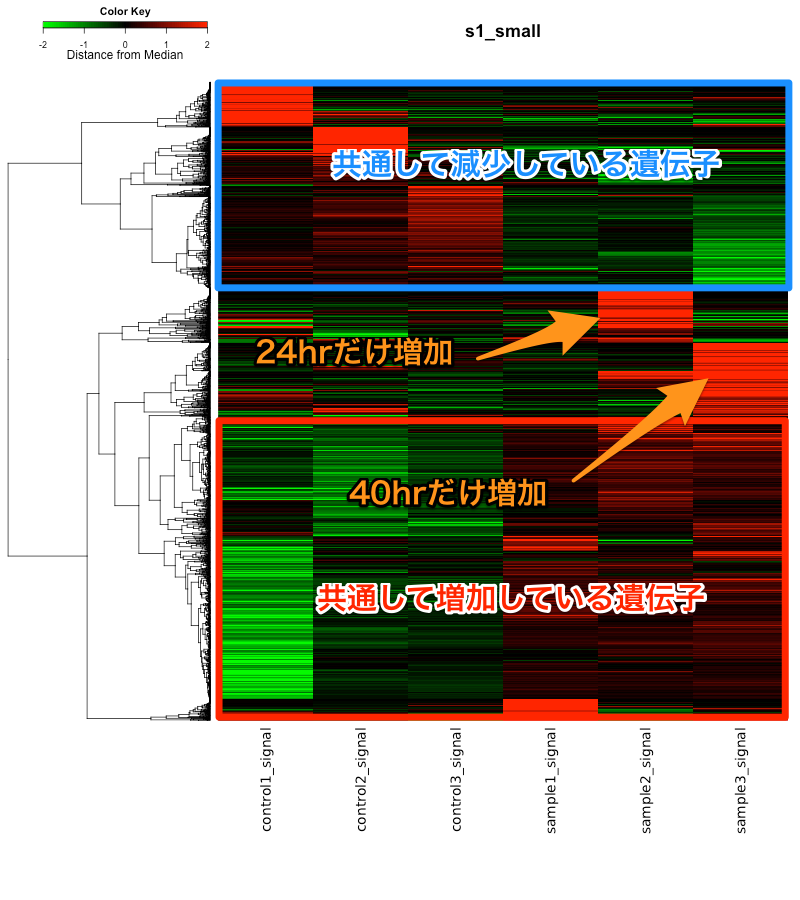
ヒートマップの結果、気になる変動パターンが見つかれば、その部分だけを取り出して、再び、DAVIDなどで確認します。そうすれば、24hrだけで増加する遺伝子に、何系の遺伝子が多いのか、判断できます。しかしながら、その結果、生物学的な意味があるのかは、個人の仮説次第です。(ヒートマップから結論が得られるわけではありません。)
参考:
{kind=link}
