eArray などのメーカーサイトからアノテーションファイル(タブ区切りのテキストファイル)をダウンロードして、ドラッグ&ドロップでエクセルで開くと、’MARCH*’ や ‘SEPT*’ などの遺伝子名は ‘*-Mar’ や ‘*-Sep’ のように自動変換されてしまいます。
(正)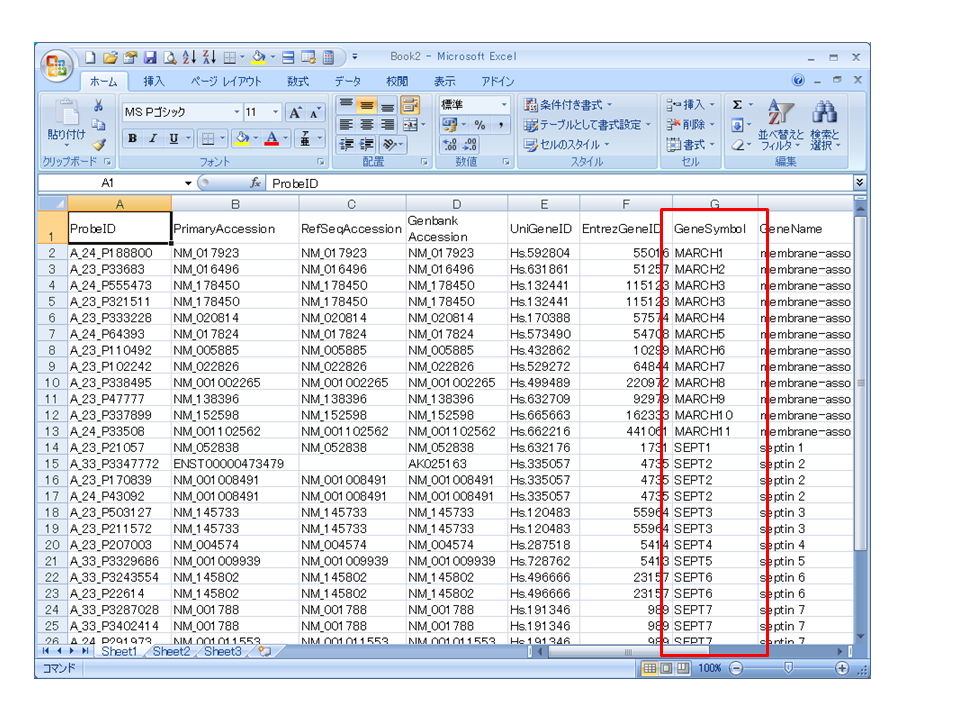
(誤)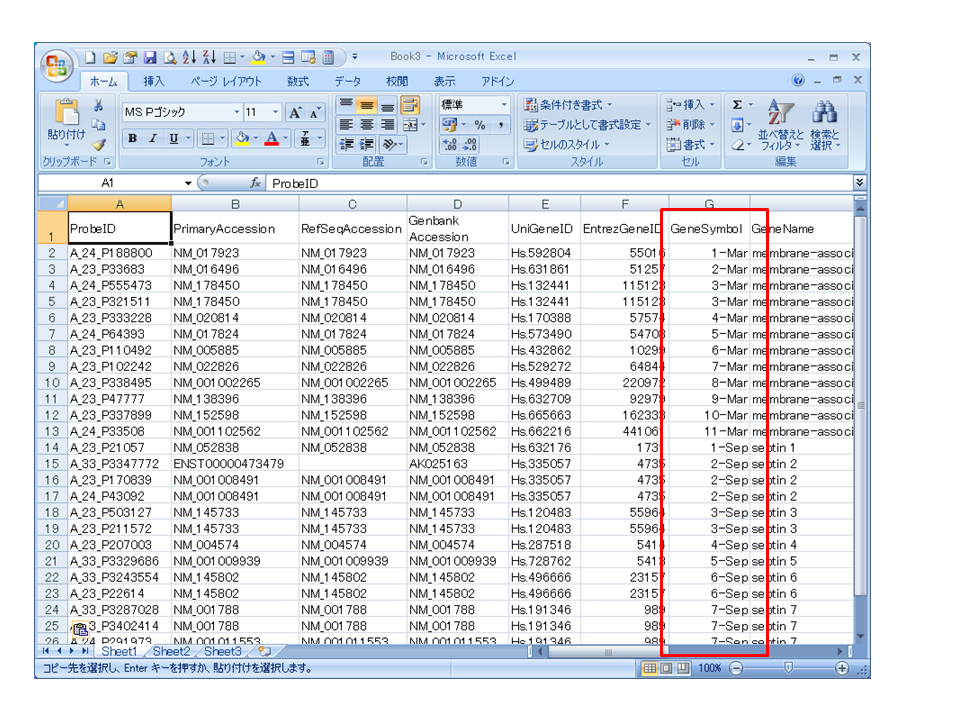
次回、この問題を回避する方法を記述します。

eArray などのメーカーサイトからアノテーションファイル(タブ区切りのテキストファイル)をダウンロードして、ドラッグ&ドロップでエクセルで開くと、’MARCH*’ や ‘SEPT*’ などの遺伝子名は ‘*-Mar’ や ‘*-Sep’ のように自動変換されてしまいます。
(正)
(誤)
次回、この問題を回避する方法を記述します。
今回も、引き続き、マイクロアレイ解析結果をLinuxコマンドを用いて簡単に操作する方法を紹介していきたいと思います。今回は、行に関する操作です。
head
ファイルの先頭を表示します。
大きいファイルの内容をちょっと確認したい場合や、ヘッダー情報を確認したい場合に使うと便利です。表示する行数も指定できます。
使い方
head test.txt # 行数オプションを指定しない場合は、5行表示します。
head -n2 test.txt > test2.txt # 先頭から2行を別ファイルに保存します。
grep
前回も出てきましたが、条件に該当する情報を抽出することができます。
行のフィルタリングに使用することができます。オプションで色んな条件を指定できます。
使い方
cat test.txt | grep “mouse” # mouseというキーワードを含む行を抽出します。
sed
代表的なテキストフィルタリングツールです。
色々な使い方ができるツールですが、今回は使用する機会の多い置換についての使い方です。
使い方
cat test.txt | sed -e “s/,/\t/g”
# カンマ(,)区切りをタブ(\t)区切りに変換します。gは1行に見つかった全てに適用の意味です。本例のようにtest.txtの全行でカンマをタブ変換したい場合は、決まった書き方で対応可能です。
サンプル>
$ cat test.txt # ターゲットとなるテキストファイル no,columnA,columnB 1,mouse,aaa 2,cat,bbb 3,mouse,ccc 4,mouse,ddd 5,cat,eee
$ head -n2 test.txt # 先頭2行だけを表示 no,columnA,columnB 1,mouse,aaa $ cat test.txt | grep "mouse" # mouseを含む行だけを抽出 1,mouse,aaa 3,mouse,ccc 4,mouse,ddd
$ cat test.txt | sed -e "s/,/\t/g" # カンマ区切りをタブ区切りに変換 no columnA columnB 1 mouse aaa 2 cat bbb 3 mouse ccc 4 mouse ddd 5 cat eee
Agilent のマイクロアレイデータを想定して、 MeV の操作方法を紹介します。
t-検定により、WT vs KO で差のある遺伝子を求める。

検定を行った上で、有意な差を持つ遺伝子群のクラスタリング図を作成。

続きます。