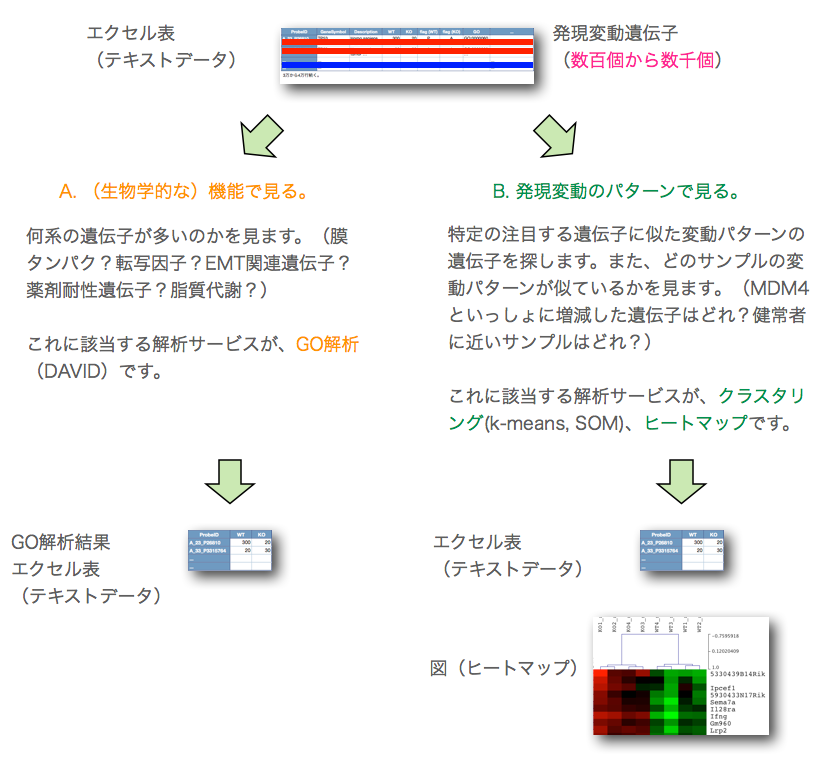
発現変動遺伝子の抽出後、まず、必要なことは、増加、減少した遺伝子(発現変動遺伝子)が、どのような遺伝子であるかを分析することです。この方法は、大きく分けて、次の2通りの方法のいずれかを用います。
A. (生物学的な)機能で見る。
B. 発現変動のパターンで見る。
A. (生物学的な)機能で見る
何系の遺伝子が多いのかを見ます。(膜タンパク?転写因子?EMT関連遺伝子?薬剤耐性遺伝子?脂質代謝?) これに該当する解析が、GO解析(DAVIDなどを使用)です。
B. 発現変動のパターンで見る
特定の注目する遺伝子に似た変動パターンの遺伝子を探します。また、どのサンプルの変動パターンが似ているかを見ます。(MDM4といっしょに増減した遺伝子はどれ?健常者に近いサンプルはどれ?) これに該当する解析が、クラスタリング(k-means, SOM)、ヒートマップです。
このA., B. の2通りの解析は、独立したものではなく、また、それだけでは終わりません。GO解析の結果をさらにヒートマップで表示して、発現変動変動のパターンを確認するケース(A. –> B.)や、特定の発現変動パターンの遺伝子を選択して、その機能をGO解析で確認するケース(B. –> A.) が通常です。それぞれの解析を単独で行っても効果的ではありません。(セルイノベーターの解析サービスでは、初めからこれらの解析サービスを含めて提供しています。)
解析はさらに続きます。

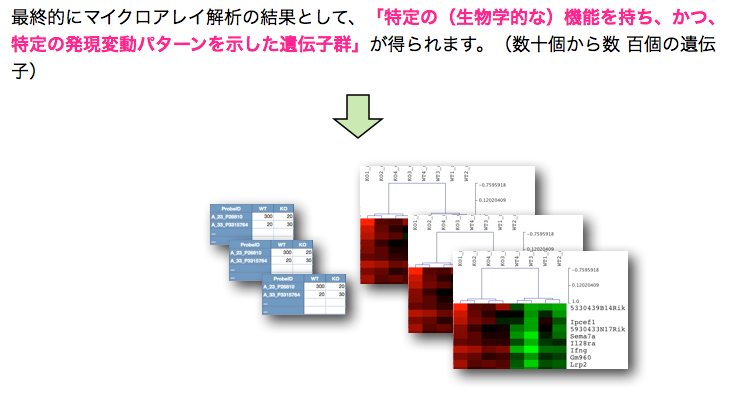
最終的にマイクロアレイ解析の結果として、「特定の(生物学的な)機能を持ち、かつ、特定の発現変動パターンを示した遺伝子群」が得られます。(数十個から数 百個の遺伝子)