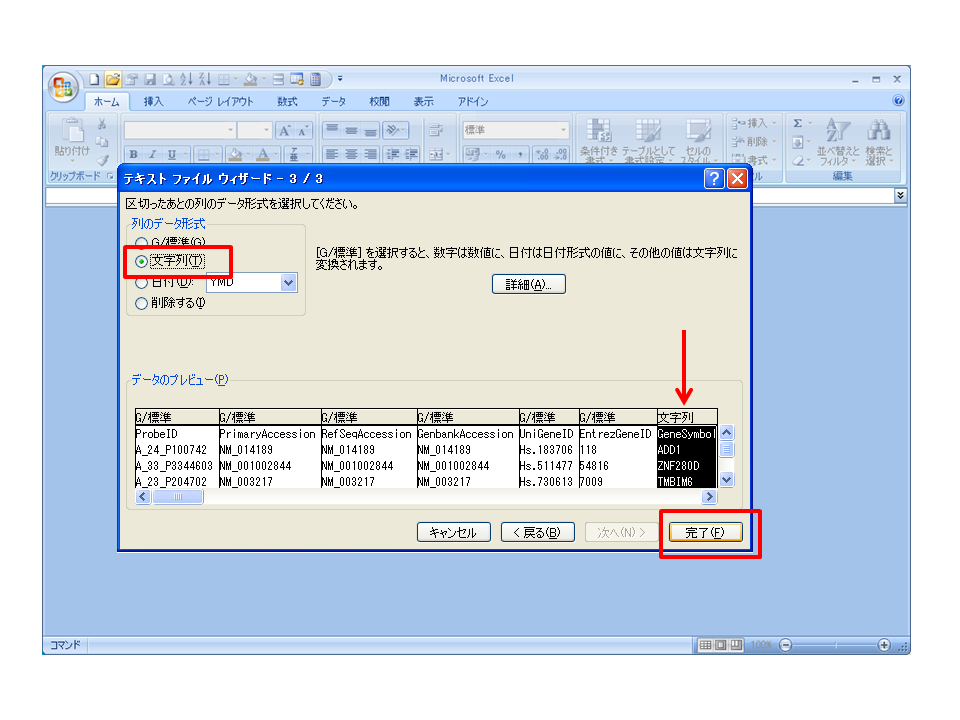
例:t-検定の結果
- 左側の “Analysis Results” に “T Tests” の結果が表示される。
- “Significant Genes” に含まれる遺伝子群が、 t-検定の結果、有意となった遺伝子群。(標準の設定では、p-value < 0.01 で有意。0.01 または 0.05 を用いることが多い。)
- サンプルデータの場合、有意となる遺伝子はない。すべての有意でない遺伝子は、 Non-significant Genes に表示される。

3.(3) 検定結果の色づけ
- 現在の色づけは、他の遺伝子と比べている(図の赤枠)。遺伝子AとBが遺伝子Cより低いため、遺伝子AとBが緑になり、遺伝子Cが赤になっている。
- 他のサンプルと比べて、シグナルの高いところは赤、低いところは緑になって欲しい。図に青枠で示されたように、遺伝子Cの6サンプル間での差を見たい。
- 色づけのための調整が必要。調整の手段は、いろいろ考えられる。各遺伝子の中央値からの距離に変換してもよいし、各遺伝子の平均値からの距離(SD何個分か)に変換してもよい。ここでは、簡単な方法として、各遺伝子の中央値からの距離に変換する方法を紹介する。

3.(4) 遺伝子ごとの中央値からの距離を求める
1) “Adjust Data -> Gene/Row Adjustments -> Median Center Genes/Rows” を選択。 この処理は取り消すことができないので注意。元のシグナル値やほかの調整を行うには、データの読み込みから、すべての作業をやり直す必要がある。
2) 再度、“Display -> Set Color Scale Limits” を選択し、色づけのスケーリングを合わせる。
- Lower Limit = -2
- Midpoint Value = 0
- Upper Limit = 2
*色の濃さは適宜、調節可能。中央値からの距離の場合、Midpoint Value は、0 以外を用いることはない。Lower = -1 , Upper Limit = 1 のように低く設定すると、色が濃くなり強調された状態となる。あまり、低い値を設定すると、どの遺伝子の差も大きいような誤解を与えるので注意。


3.(5) t-検定結果の保存
クラスタリング図を右クリックして出るメニューから、 “Save cluster…” を選択すると、画像ファイルとして結果を保存できる。