8. 解析結果1:アノテーションの解析結果を見る例です。ここでは、”Functional
Annotation Clustering” を見る方法を解説します。中程の ”Functional Annotation Clustering” のボタンをクリックします。

新しいウィンドウに解析結果が表示されます。Functional Annotation Clustering では、同じような機能を持った遺伝子群を1つのクラスターとして考え、スコアの高いクラスターの順に表示されます。1つのクラスターには、metabolic process, anion transport などの Gene Ontology (GO) が複数含まれています。アップロードした遺伝子リスト中、それぞれの GO をアノテーションに持つ遺伝子は、GO の隣の青いバーをクリックすることで見ることができます(Gene Report)。 P_Value は、0.05 (5.0E-2)以下が統計的に有意と判断される目安です。
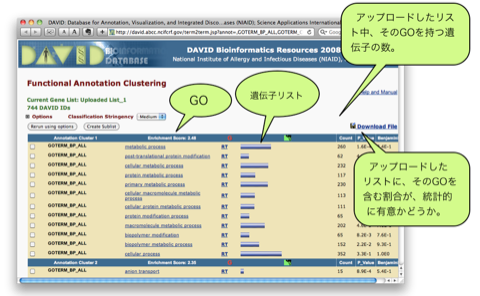
青いバーをクリックして表示される Gene Report は、右上の “Download file” をクリックすることで、ダウンロードできます。
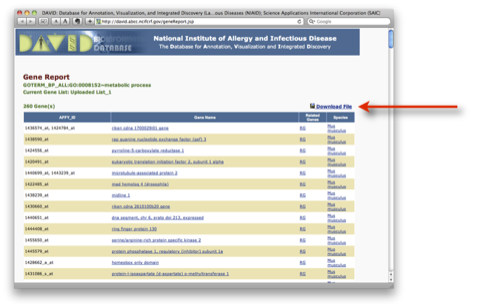
ダウンロードした Gene Report は、Excel で読み込むことができます。開くときに、選択対象を ”すべてのファイル” とします (“すべての読み込み可能なファイル”ではなく)。
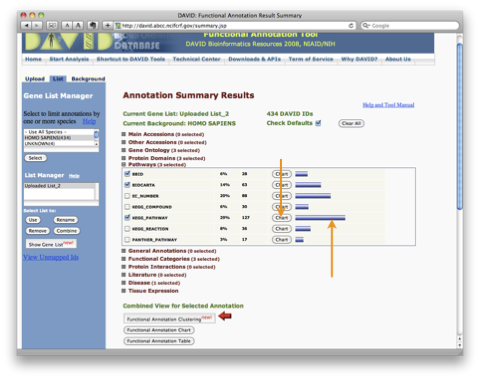
9. 解析結果2:パスウェイを表示させる例です。 Annotation Summary Results の画面(7. の画面)の “Pathways (3 selected)” を選択します。
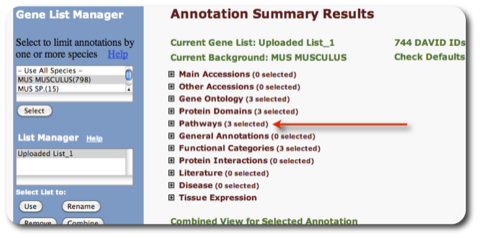
同じウィンドウの中にアノテーション情報として登録されているパスウェイデータベースの一覧が表示されます。パスウェイデータベースの横の数字は、 アップロードした遺伝子リストのうち、パスウェイデータベースにヒットした割合と個数です。 “Chart” ボタンをクリックすると、 “Functional Annotation Chart” のウィンドウが表示されます。また、Chart ボタンの隣の ”青いバー” をクリックすると、 ”Functional Annotation Table” のウィンドウが表示されます。さらに、それぞれのウィンドウにおいて、各パスウェイ名をクリックすると、パスウェイの画像を表示できます。
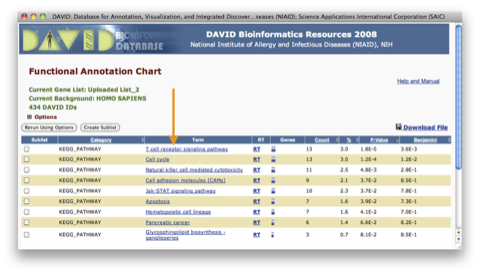
パスウェイデータベース(ここでは KEGG )に登録されている ”Apoptosis” や ”Cell Cycle” といった個々のパスウェイの中から、アップロードした遺伝子リストに含まれる遺伝子が載っている(マップされる)パスウェイを探すには、 ”Functional Annotation Chart” を参照します。スコアに関係なく、マップされるパスウェイを知りたい場合は、”Functional Annotation Table” を参照します。
Functional Annotation Chart ウィンドウには、アップロードした遺伝子リスト中の遺伝子を、統計的に有意な割合で含むパスウェイだけが表示されます。 “Term” 中の各パスウェイ名をクリックするとパスウェイが表示されます。
Functional Annotation Table ウィンドウには、アップロードした遺伝子リストのうち、パスウェイデータベースにヒットした遺伝子の一覧が表示されます。各パスウェイ名をクリックするとパスウェイが表示されます。パスウェイの数が多い場合は、ブラウザの検索機能を利用すると便利です。

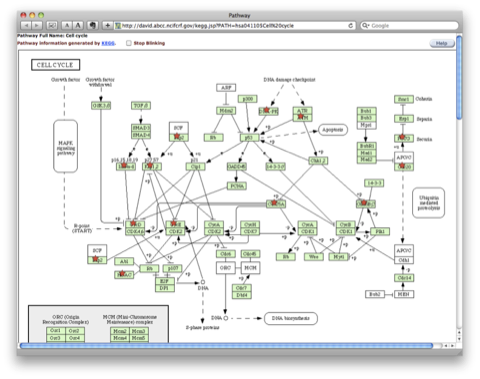
マップされたパスウェイの例:下図は、”Cell Cycle” のパスウェイにマップされた場合の例です。アップロードした遺伝子リストに含まれていた遺伝子は、星印をつけて表示されます。各遺伝子をクリックすると、遺伝子について詳細な情報を見ることができます。
Reference
Huang et al. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols (2009) vol. 4 (1) pp. 44-57.



{kind=link}