データを読み込むところまでを紹介します。フォーマットを合わせることができれば、操作は難しくありません。(データは、3. で示すフォーマットで事前に準備しておく必要があります。)
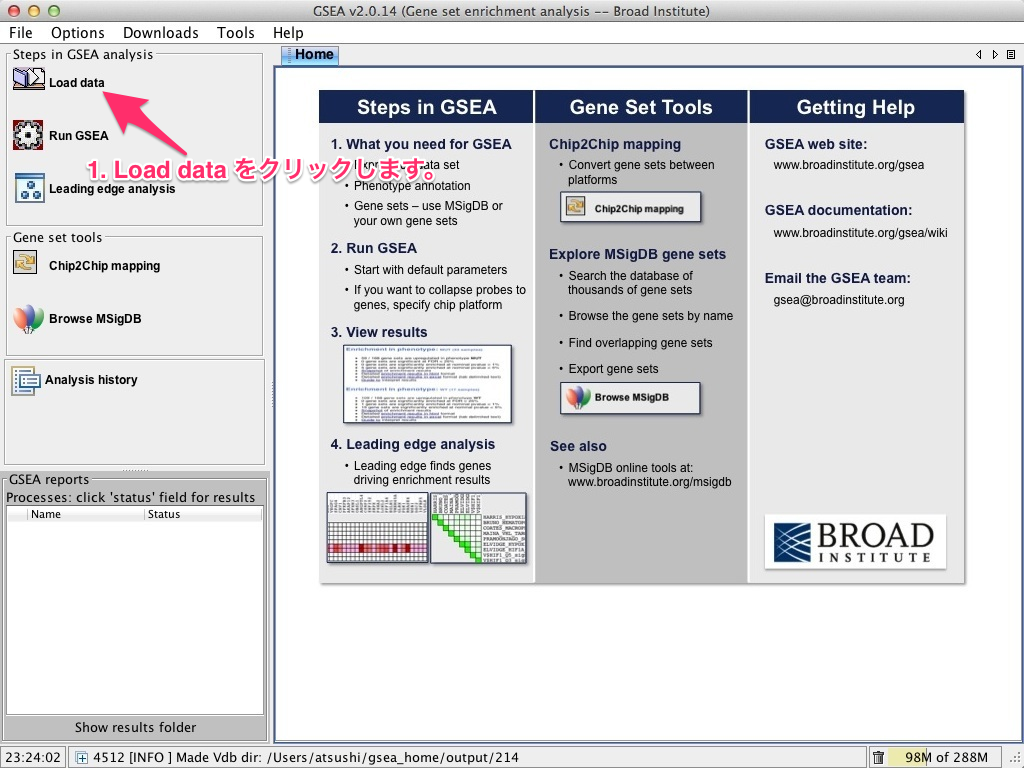
1. Load data をクリック
メインとなるウィンドウの左上にある “Load data” のボタンをクリックします。
2. 読み込むファイルの選択
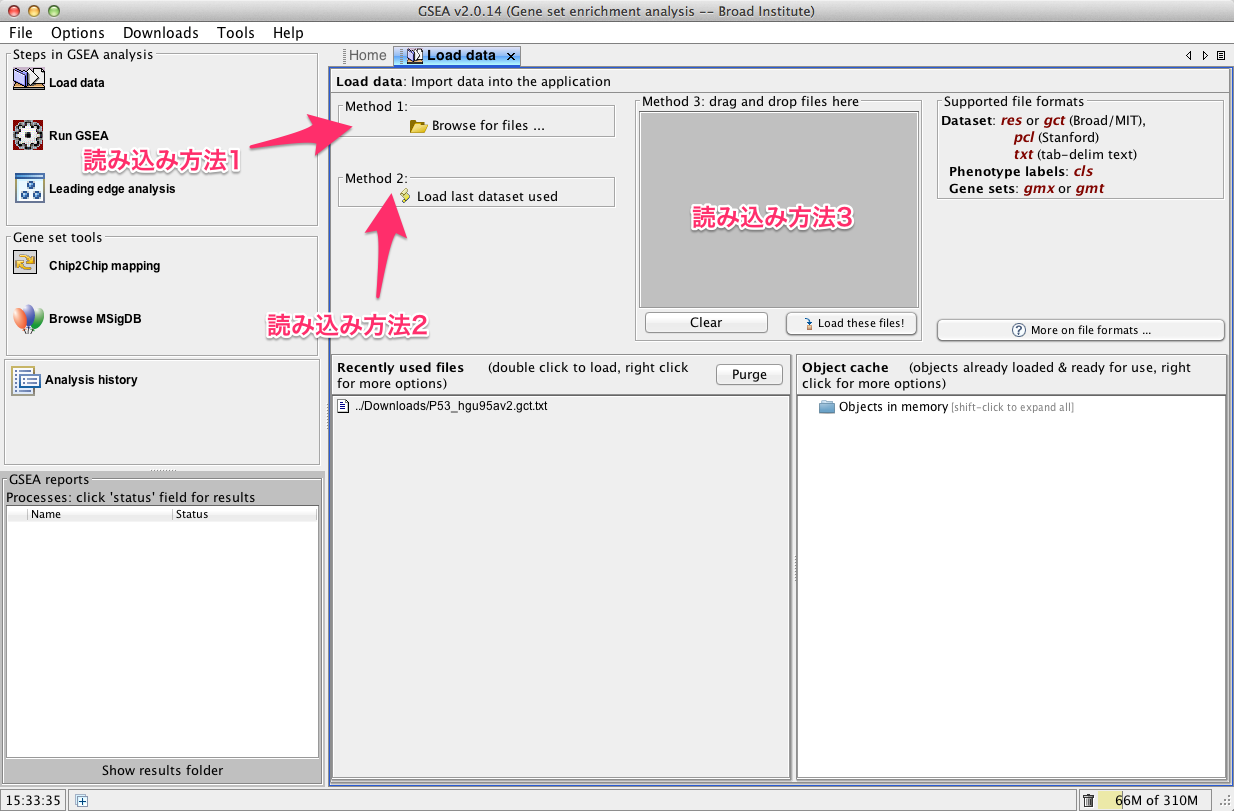
右側にデータを読み込むためのタブが表示されます。読み込む方法は3種類あります。いずれかの方法を使用します。通常は、読み込み方法1を使うとよいでしょう。
- 読み込み方法1: ボタンをクリックすると表示されるダイアログからファイルを選択します。
- 読み込み方法2: 過去に開いたファイルの一覧から指定します。
- 読み込み方法3: ファイルをドラッグ&ドロップして、右下の “Load these files!” をクリックします。
3. ロードできるデータのフォーマット
読み込むことができるファイルの形式(フォーマット)は、GCT, RES, PCL, TXT の4種類です。一般的には、 GCT フォーマットを利用するのがよいでしょう。各フォーマットの書式については、こちらに書かれています。例は、Affymetrix のデータですが、Agilent の場合も GCT フォーマットに合わせれば読み込むことができます。(RESフォーマットであれば、Agilent のフラグ A, M, P を読み込ませることができますが、現在、このフラグの情報は無視されるようです。)
なお、読み込むデータは、あらかじめ数値化や正規化しておく必要があります。CELファイルや Feature Extraction のファイルなど、raw データをそのまま読み込むことはできません。
正規化後のデータをエクセルなどで読み込んで、必要な書式に整形します。テキスト形式のデータをエクセルに読み込む場合は、遺伝子名が変換されてしまわないように注意してください(前記事参照)。
GCTフォーマットに必要なポイント
- ポイント1: 1行目に #1.2 と書く。これはそういう決まりらしいです。
- ポイント2: 2行目の1列目に、遺伝子数(プローブ数)を書きます。あらかじめカウントしておきましょう。
- ポイント3: 2行目のプローブ数の横に、サンプル数を書きます。
- ポイント4: 1列目は、NAME として、 ProbeID (ProbeSetID) を指定します。重複するIDであってはいけません。遺伝子名ではダメです。
- ポイント5: 2列目は Description (=説明書き)です。空欄にはできません。何も情報がなければ、 “na” と書きます。
- ポイント6: 3列目からシグナル値が始まるようにします。サンプル名は重複してはいけません。unlog (natural scale) でも log変換後 (log scale) でもよいらしいです。
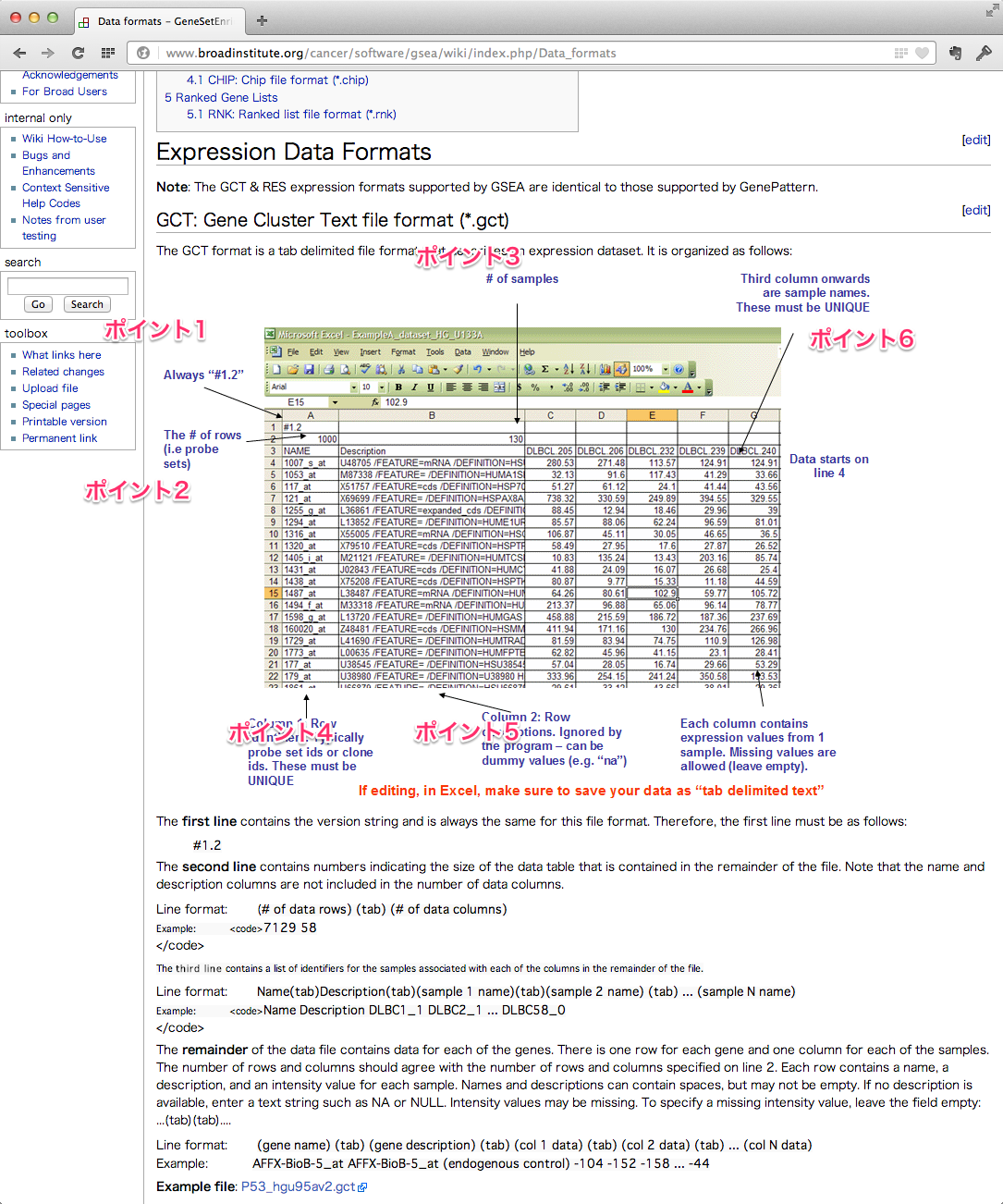
最後にファイルを「タブ区切りのテキスト形式」で保存します。エクセルの形式(.xls, .xlsx)では読み込むことはできません。
4. ファイルの読み込み
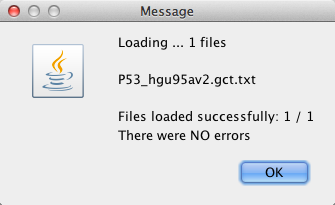
作成しておいた GCT フォーマットのファイルを、2. のいずれかの方法で選択すると、GSEA にファイルが読み込まれます。正常に読み込まれると、下図のように NO errors と表示されます。読み込んだ後は、GSEA の実行に必要なパラメーターの設定へと進みます(次回)。